Omix - Pseudotemporal multi-omics integration
This vignette was created by Eléonore Schneegans and Nurun Fancy
10 April, 2024
Omix_vignette_MAP_CV_pseudotemporal.RmdOverview
The goal of Omix is to provide tools in R to build a complete analysis workflow for integrative analysis of data generated from multi-omics platform.
Generate a multi-omics object using
MultiAssayExperiment.Quality control of single-omics data.
Formatting, normalisation, denoising of single-omics data.
Separate single-omics analyses.
Integration of multi-omics data for combined analysis.
Publication quality plots and interactive analysis reports based of shinyApp.
Currently, Omix supports the integration of bulk transcriptomics and bulk proteomics.
Running Omix
The Omix pipeline requires the following input:
rawdata_rna: A data-frame containing raw RNA count with rownames as gene and colnames as samples.rawdata_protein: A data-frame containing protein abundance with rownames as proteins and colnames as samples.map_rna: A data-frame of two columns namedprimaryandcolnamewhere primary should contain unique sample name with a link to sample metadata and colname is the column names of therawdata_rnadata-frame.map_protein: A data-frame of two columns namedprimaryandcolnamewhere primary should contain unique sample name with a link to sample metadata and colname is the column names of therawdata_proteindata-frame.metadata_rna: A data-frame containingrnaassay specific metadata where rownames are same as the colnames ofrawdata_rnadata.frame.metadata_protein: A data-frame containingproteinassay specific metadata where rownames are same as the colnames ofrawdata_proteindata.frame.individual_metadata: A data-frame containing individual level metadata for both omics assays.
Call required packages for vignette:
First we must load the data from Omix for the vignette:
outputDir <- tempdir()
ctd_fp <-file.path(outputDir, "ctd.rds")
ensembl_fp <-file.path(outputDir, "ensembl.rds")
TF_fp <- file.path(outputDir, "Enrichr_Queries.gmt.txt")
GO_fp <- file.path(outputDir, "GO_Biological_Process_2021.txt")
download.file(url = "https://raw.githubusercontent.com/eleonore-schneeg/OmixData/main/ctd-2.rds",destfile=ctd_fp)
download.file(url = "https://raw.githubusercontent.com/eleonore-schneeg/OmixData/main/ensembl_mappings_human.tsv",destfile=ensembl_fp)
download.file(url = "https://raw.githubusercontent.com/eleonore-schneeg/OmixData/main/Enrichr_Queries.gmt.txt",destfile=TF_fp)
download.file(url = "https://raw.githubusercontent.com/eleonore-schneeg/OmixData/main/GO_Biological_Process_2021.txt",destfile=GO_fp)
synapser::synLogin(email=Sys.getenv("SYNAPSE_ID"), authToken=Sys.getenv("AUTHTOKEN"))Welcome, ems2817!NULL
rawdata_rna <- read.csv(synapser::synGet('syn51516100')$path, header=T, stringsAsFactors = F, row.names=1)
rawdata_protein <- read.csv(synapser::synGet('syn51516105')$path, header=T, stringsAsFactors = F, row.names=1)
map_rna <-read.csv(synapser::synGet('syn51516102')$path, header=T, stringsAsFactors = F, row.names=1)
map_prot <- read.csv(synapser::synGet('syn51516104')$path, header=T, stringsAsFactors = F, row.names=1)
metadata_rna<-read.csv(synapser::synGet('syn51516103')$path, header=T, stringsAsFactors = F, row.names=1)
metadata_prot <- read.csv(synapser::synGet('syn51516106')$path, header=T, stringsAsFactors = F, row.names=1)
individual_metadata <-read.csv(synapser::synGet('syn51516101')$path, header=T, stringsAsFactors = F, row.names=1)
clusters<-readRDS(synapser::synGet('syn51516107')$path)
integrative_model<-readRDS(synapser::synGet('syn51520491')$path)
ctd <- readRDS(paste0(outputDir,'/ctd.rds'))
ensembl <-read.delim(file=paste0(outputDir,'/ensembl.rds'), sep = '\t', header = TRUE)
rna_qc_data_matrix <- NULLSanity check
## [1] TRUE## [1] TRUERaw rna counts
rawdata_rna is a data-frame of raw counts, with features
as rows and samples as columns
print(rawdata_rna[1:5,1:5])## IGF117745 IGF117748 IGF117756 IGF117758 IGF117759
## ENSG00000223972 0 0 1 0 1
## ENSG00000227232 23 26 40 26 30
## ENSG00000278267 7 5 1 4 0
## ENSG00000243485 0 0 0 1 0
## ENSG00000284332 0 0 0 0 0Raw protein abundance
rawdata_protein is a data-frame of raw protein
abundances, with features as rows and samples as columns
print(rawdata_protein[10:15,10:15])## BBN_16213_SOM_P2WH4_001 BBN_18399_MTG_P2WF5_001
## MAP1LC3B;MAP1LC3B2 140.21600 118.1090
## PGP 33.91880 24.3759
## BTBD17 NA NA
## WIPF3 39.10300 98.0357
## IGLON5 8.55527 10.2548
## TUBAL3 9432.14000 10277.5000
## BBN_18399_SOM_P2WF6_001 BBN_18813_SOM_P3WB2_001
## MAP1LC3B;MAP1LC3B2 116.8410 109.55500
## PGP 15.2674 31.62250
## BTBD17 13.8635 NA
## WIPF3 68.9909 26.42480
## IGLON5 21.7657 8.52512
## TUBAL3 9565.3200 10144.50000
## BBN_19214_MTG_P2WA3_001 BBN_19214_SOM_P2WA4_001
## MAP1LC3B;MAP1LC3B2 121.1260 166.2360
## PGP 18.2978 26.0274
## BTBD17 NA 17.6870
## WIPF3 58.8438 NA
## IGLON5 12.5892 20.8584
## TUBAL3 9069.5600 10454.6000Maps
Maps are data frame containing two columns: primary and
colname. The primary column should be the
individual ID the individual metadata, and the colname the
matched sample names from the raw matrices columns. If sample and
individual ids are the same, maps aren’t needed (primary and colnames
are the same).
## primary colname
## 1 BBN_10099-MTG BBN_10099_MTG_P3WH10_001
## 2 BBN_10099-SOM BBN_10099_SOM_P4WA1_001
## 3 BBN_10109-MTG BBN_10109_MTG_P4WA3_001
## 4 BBN_10109-SOM BBN_10109_SOM_P4WA4_001
## 5 BBN_10611-MTG BBN_10611_MTG_P2WD10_001
## 6 BBN_10611-SOM BBN_10611_SOM_P2WE1_001## primary colname
## 1 BBN002.30035-SOM IGF117745
## 4 BBN_7519-SOM IGF117748
## 12 BBN003.32235-MTG IGF117756
## 14 BBN_21792-SOM IGF117758
## 15 BBN_7617-MTG IGF117759
## 16 BBN_9984-SOM IGF117760Technical metadata
Technical metadata are data-frames contain the column
colname that should match the sample names in the raw
matrices, and any additional columns related to technical artefacts like
batches
## sample_id sample_name RIN Amyloid_Beta pTau
## IGF117745 BBN002.30035-SOM IGF117745 6.4 0.343451 0.00553998
## IGF117748 BBN_7519-SOM IGF117748 8.1 NA NA
## IGF117756 BBN003.32235-MTG IGF117756 2.7 2.027670 6.98931000
## IGF117758 BBN_21792-SOM IGF117758 5.3 1.529590 0.04699940
## IGF117759 BBN_7617-MTG IGF117759 2.0 3.262270 2.55134000
## IGF117760 BBN_9984-SOM IGF117760 7.7 3.653590 1.65894000
## pct_pTau_Positive PHF1
## IGF117745 0.0426309 0.00121131
## IGF117748 NA NA
## IGF117756 5.2597500 2.42065000
## IGF117758 0.1894760 0.00342135
## IGF117759 0.3749760 1.56408000
## IGF117760 1.5665900 0.06801730## sample_id sample_name batch
## BBN_10099_MTG_P3WH10_001 BBN_10099-MTG BBN_10099_MTG_P3WH10_001 P3
## BBN_10099_SOM_P4WA1_001 BBN_10099-SOM BBN_10099_SOM_P4WA1_001 P4
## BBN_10109_MTG_P4WA3_001 BBN_10109-MTG BBN_10109_MTG_P4WA3_001 P4
## BBN_10109_SOM_P4WA4_001 BBN_10109-SOM BBN_10109_SOM_P4WA4_001 P4
## BBN_10611_MTG_P2WD10_001 BBN_10611-MTG BBN_10611_MTG_P2WD10_001 P2
## BBN_10611_SOM_P2WE1_001 BBN_10611-SOM BBN_10611_SOM_P2WE1_001 P2Individual metadata
individual_metadata should contain individual level
metadata, where one column matches the primary column in
maps.
## sample_id brain_region age BBN_ID sex diagnosis apoe_final Braak PMD
## 1 BBN_10099-MTG MTG 91 BBN_10099 F Control E3/E3 2 22
## 2 BBN_10099-SOM SOM 91 BBN_10099 F Control E3/E3 2 22
## 3 BBN_10109-MTG MTG 80 BBN_10109 F Control E2/E3 2 23
## 4 BBN_10109-SOM SOM 80 BBN_10109 F Control E2/E3 2 23
## 5 BBN_10611-MTG MTG 92 BBN_10611 F AD E3/E3 3 30
## 6 BBN_10611-SOM SOM 92 BBN_10611 F AD E3/E3 3 30
## amyloid pTau PHF1 AD MTG SOM
## 1 0.585285 0.00649899 0.00168676 0 1 0
## 2 0.226991 0.00960187 NA 0 0 1
## 3 0.782684 0.92852500 0.67876900 0 1 0
## 4 0.059688 0.00193356 NA 0 0 1
## 5 0.307882 0.01312750 0.01797600 1 1 0
## 6 0.530825 0.00562307 0.00824549 1 0 1Step one - Generate a MultiAssayExperiment object
To run Omix, we first need to generate a multi-omics object The package currently supports transcriptomics and proteomics bulk data only.
individual_metadata$diagnosis <- factor(individual_metadata$diagnosis,
levels = c("Control", "AD"))
individual_metadata$sex <- factor(individual_metadata$sex)
multiomics_object=generate_multiassay(rawdata_rna =rawdata_rna,
rawdata_protein = rawdata_protein,
individual_to_sample=FALSE,
map_rna = map_rna,
map_protein = map_prot,
metadata_rna = metadata_rna,
metadata_protein = metadata_prot,
individual_metadata = individual_metadata,
map_by_column = 'sample_id',
rna_qc_data=FALSE,
rna_qc_data_matrix=NULL,
organism='human')## ✔ Ensembl ID conversion to gene symbol## ✔ Retrieval of gene biotype## ✔ RNA raw data loaded## class: SummarizedExperiment
## dim: 58884 48
## metadata(1): metadata
## assays(1): rna_raw
## rownames(58884): ENSG00000223972 ENSG00000227232 ... ENSG00000277475
## ENSG00000268674
## rowData names(3): ensembl_gene_id gene_name gene_biotype
## colnames(48): IGF117745 IGF117748 ... IGF117836 IGF117837
## colData names(7): sample_id sample_name ... pct_pTau_Positive PHF1## ✔ Protein raw data loaded## class: SummarizedExperiment
## dim: 3228 82
## metadata(1): metadata
## assays(1): protein_raw
## rownames(3228):
## IGKV2-28;IGKV2-29;IGKV2-30;IGKV2-40;IGKV2D-26;IGKV2D-28;IGKV2D-29;IGKV2D-30;IGKV2D-40
## IGKV3-11;IGKV3D-11 ... FAM169A SEC23IP
## rowData names(1): gene_name
## colnames(82): BBN_10099_MTG_P3WH10_001 BBN_10099_SOM_P4WA1_001 ...
## BBN003.35520_MTG_P2WD4_001 BBN003.35521_SOM_P2WD5_001
## colData names(3): sample_id sample_name batch## ✔ MultiAssayExperiment object generated!The MultiAssayExperiment object was succesfully created. The
following steps will process and perform QC on each omic layers of the
multiomics_object object.
Step two - Process transcriptomics data
multiomics_object=process_rna(multiassay=multiomics_object,
transformation='rlog',
protein_coding=TRUE,
min_count = 10,
min_sample = 0.5,
dependent = "diagnosis",
levels = c("Control","AD"),
covariates=c('age','sex','PMD'),
filter=TRUE,
batch_correction=TRUE,
batch=NULL,
remove_sample_outliers= FALSE)## ✔ NORMALISATION & TRANSFORMATION## ✔ GENE FILTERING## ✔ Keeping only protein coding genes## ✔ 39155 / 58884 non protein coding genes were dropped## ✔ 19729 protein coding genes kept for analysis## ✔ QC parameters selected require genes to have at least 50 % of samples with counts of 10 or more## ✔ 4849 / 19729 genes were dropped## ✔ 14880 genes kept for analysis## ✔ RLOG TRANSFORMATION## ✔ BATCH CORRECTION## ✔ Using Limma on to remove technical artefacts, and age as biological confoundersUsing Limma on to remove technical artefacts, and sex as biological confoundersUsing Limma on to remove technical artefacts, and PMD as biological confounders## ✔ Transcriptomics data processed!## ✔ Processing parameters saved in metadataStep three - Process proteomics data
multiomics_object=process_protein(
multiassay=multiomics_object,
filter=TRUE,
min_sample = 0.5,
dependent = "diagnosis",
levels = c("Control","AD"),
imputation = 'minimum_value',
remove_feature_outliers= FALSE,
batch_correction= FALSE,
batch="batch",
correction_method="median_centering",
remove_sample_outliers=FALSE,
denoise=TRUE,
covariates=c('PMD','sex','age'))## ✔ SCALING NORMALIZATION## ✔ FILTERING## ✔ 283 / 3228 proteins filtered## ✔ 2945 proteins kept for analysis## ✔ IMPUTATION## ✔ Imputation of remaining missing values based on
## 50% of minimum level of abundance for each protein## ✔ DENOISING BIOLOGICAL COVARIATES## ✔ Using Limma on PMD as biological confoundersUsing Limma on sex as biological confoundersUsing Limma on age as biological confoundersStep four - Vertical integration
Omix supports a range of vertical integration models:
Possible integration methods are
MOFA,DIABLO,sMBPLS,iCluster,MEIFESTOThe choice of the integration models depends on the research use case of interest.
Here we display the use of Omix on a popular integration method, MOFA or Multi-Omics Factor analysis (Argelaguet et al. 2018).
The
Meifestomodel is an extension ofMOFAto enable time as a covariate.
In this vignette, we proceed with a pseudotemporal multi-omics integration of 40 brain Alzheimer’s disease (AD) and Control samples coming from two brain regions
- The somatosensory cortext (SOM)
- The middle frontal gyrus (MTG)
The MTG is known to be affected earlier during AD progression, while SSC at later stages. Using these two regions as pseudotemporal proxi in the integrative process, we are able to gain a deeper understanding of biological mechanisms that occur during AD progression.
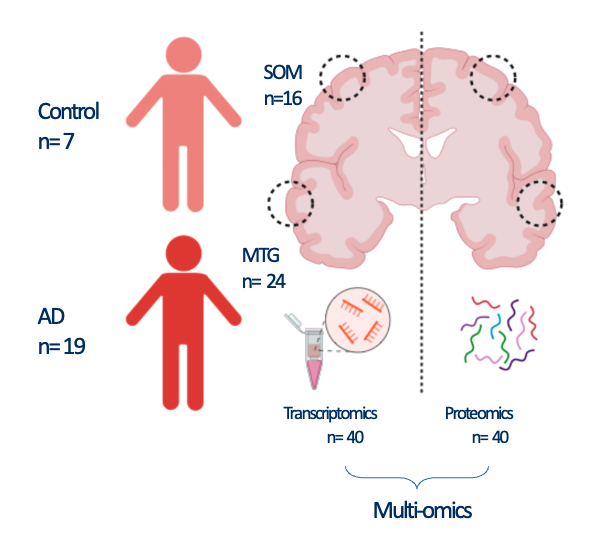
Pseudotemporal multi-omics integration using MOFA
multiomics_object=vertical_integration(multiassay=multiomics_object,
slots = c(
"rna_processed",
"protein_processed"
),
integration='MOFA',
ID_type = "gene_name",
dependent='diagnosis',
intersect_genes = FALSE,
num_factors = 15,
scale_views = FALSE,
most_variable_feature=TRUE)The integration process is successful and all integration related
object are stored in the integration slot of the
multi-omics object.
print(multiomics_object@metadata$integration$MOFA)## Trained MOFA with the following characteristics:
## Number of views: 2
## Views names: mRNA proteins
## Number of features (per view): 2945 2945
## Number of groups: 1
## Groups names: group1
## Number of samples (per group): 40
## Number of factors: 15Step five - Post integration downstream analyses
Omix provides a range of built-in downstream
analyses functions and visualisations. All downstream analyses will be
performed on the integrated object stored in the integrated
slot.
Load integrated object
integrated_object=multiomics_object@metadata$integration$MOFA
metadata=integrated_object@samples_metadataFactor explorations
Correlation of factors with covariates
integrated_object@samples_metadata$SOM=ifelse(integrated_object@samples_metadata$brain_region=='SOM',1,0)
plot=correlation_heatmap(integrated_object,
covariates=c("AD","Braak",'MTG','SOM',
'amyloid','pTau','PHF1'))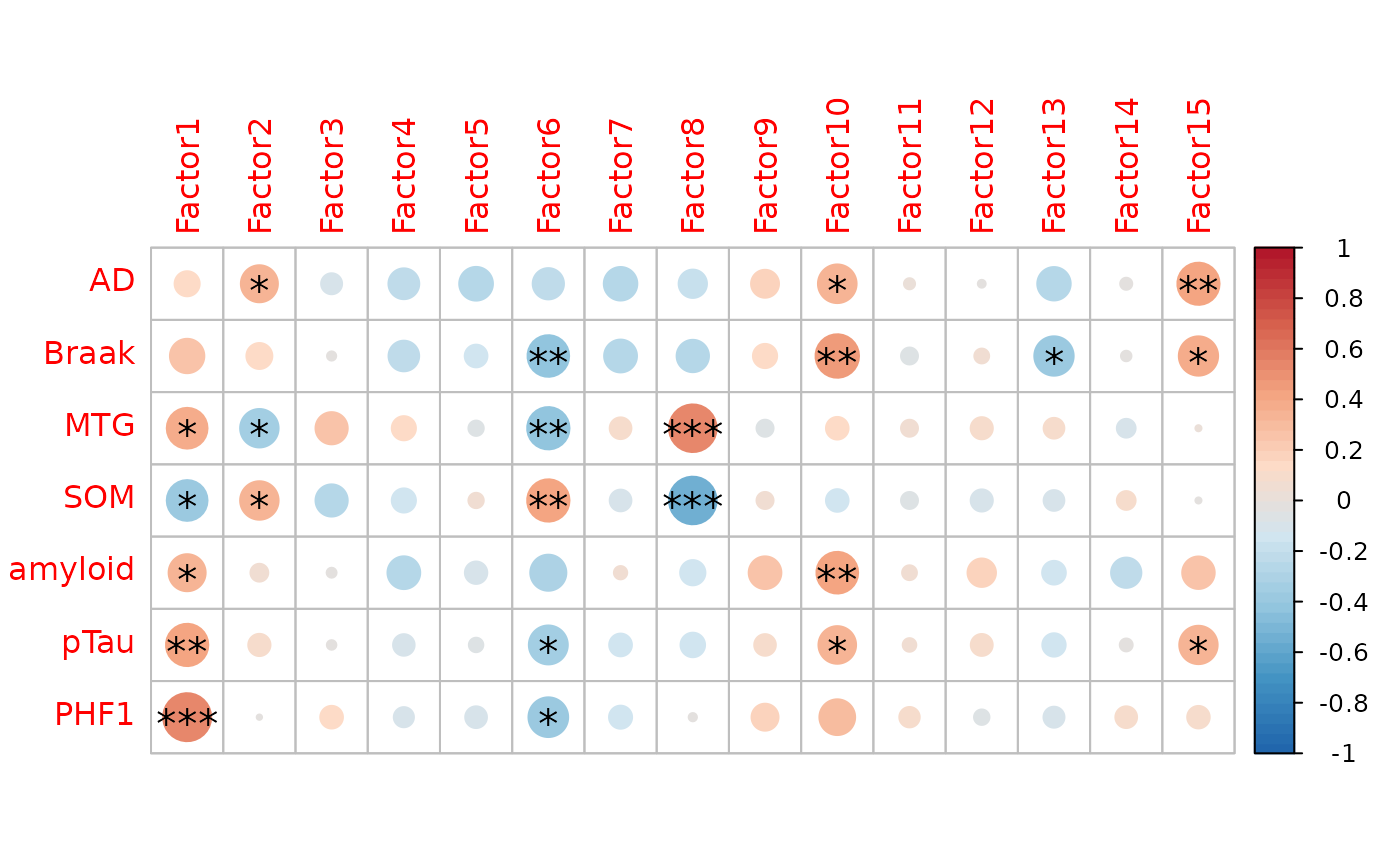
Factors 6 and 8 will be chosen for downstream analysis:
Factor 6: Is strongly correlated with neuropathological variables including PHF1 and amyloid.
Factor 8: Is strongly correlated with pseudotemporal covariates, MTG and SOM
Both share some level of variance explained by the transcriptomics and proteomics layers
Consequentially, If we project samples on these two axis of variation, we can infer on the pseudotemporal trajectory of neuropathology at the RNA and protein level.
Integrated latent space
Chosen integrated axes of variation
MOFA2::plot_factors(integrated_object,
factors=c(8,6),
color_by='PHF1', shape_by = 'brain_region', scale = T)+
xlab('Factor 8 ~ Pseudotime' )+
ylab('Factor 6 ~ Neuropathology' ) 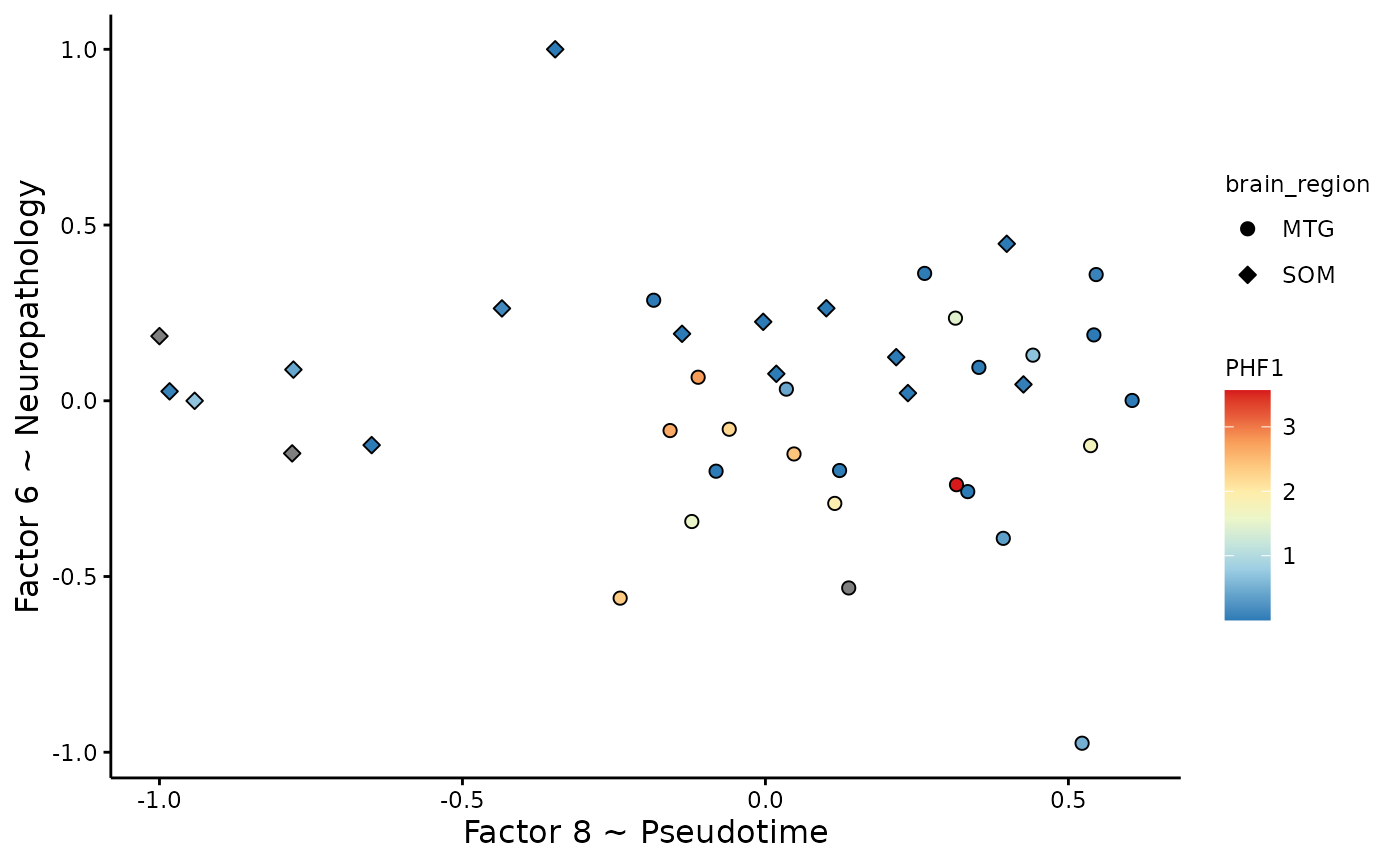
Pseudotime inference with Slingshot
The goal of slingshot (Street et al. 2017), is to use
clusters of cells to uncover global structure and convert this structure
into smooth lineages represented by one-dimensional variables, called
“pseudotime.”
Here, we extend the use of
slingshotto infer on individual level pseudotime to infer on Alzheimer’s disease trajectory.
slingshot takes a matrix of embeddings in reduced
dimension as input, which here are Factors 8 and 6 from the
MOFA multi-omics integration. We can optionally specify the
cluster to start or end the trajectory based on biological knowledge.
Here, the starting cluster should be 1 as it coincides with
lowest neuropathology levels and most “recent” affected region in terms
of AD trajectory.
#clusters=MOFA2::cluster_samples(integrated_object,
# k=3,
# factors = c(8,6))
#load clusters for reproducibility
MOFA2::plot_factors(integrated_object,
factors=c(8,6),
color_by=clusters$cluster, scale = F)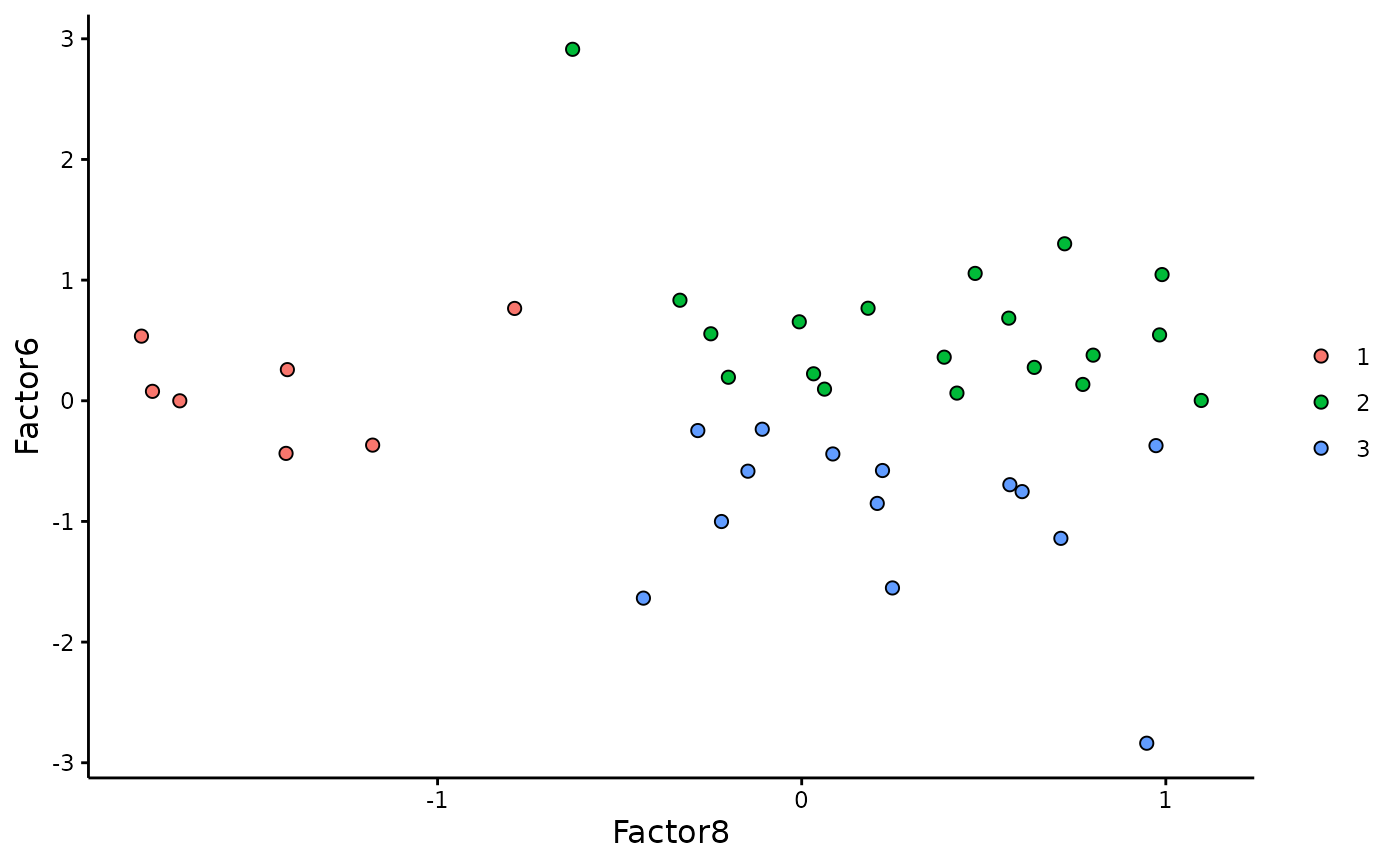
pseudotime=pseudotime_inference(integrated_object,
clusters,
time_factor=8,
second_factor=6,
start.clus = 1,
end.clus=3,
lineage = 'Lineage1')
pseudotime$plot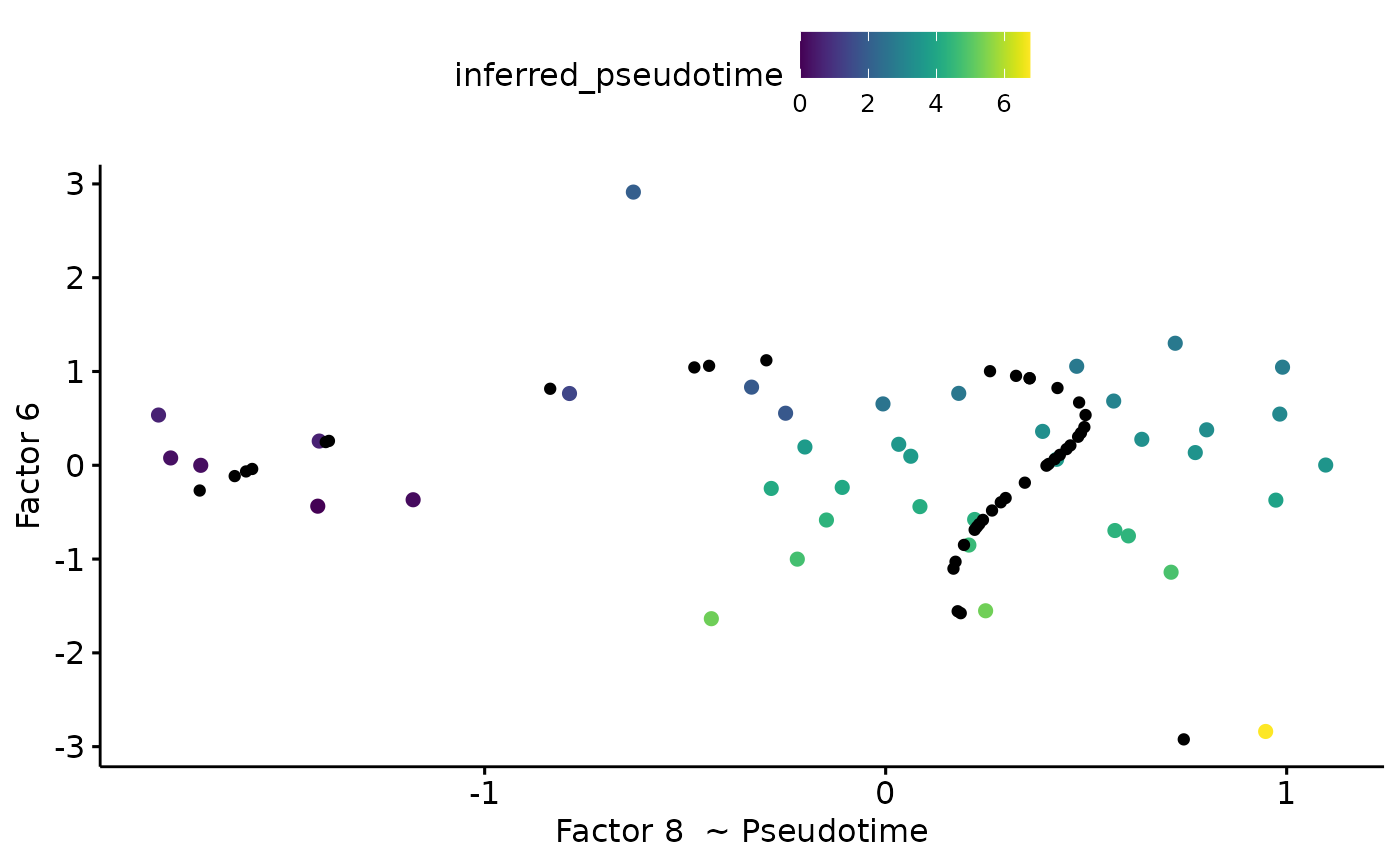
Update the integrated_object with the pseudotime
multiomics_object@metadata$integration$MOFA=pseudotime$model
integrated_object=multiomics_object@metadata$integration$MOFA
MOFA2::samples_metadata(integrated_object)$inferred_pseudotime## [1] 4.3991398 0.0000000 3.2695341 0.6197033 3.3734278 3.5719823 3.1405570
## [8] 3.3321288 3.5080573 3.6197082 1.4029378 3.6919371 2.0234374 2.7175514
## [15] 2.5931510 4.3672023 1.8283014 5.2932327 4.7431202 3.4678981 4.3375355
## [22] 0.2645565 4.5626404 0.6050538 1.8689173 2.8424525 2.7175514 4.2916315
## [29] 2.6748828 4.0987023 0.1769606 4.8168531 3.0058323 3.6727096 6.7492822
## [36] 3.8811122 0.2341031 5.2714195 4.1889176 4.0515236Extract features that are driving Factor 6 based on feature weights
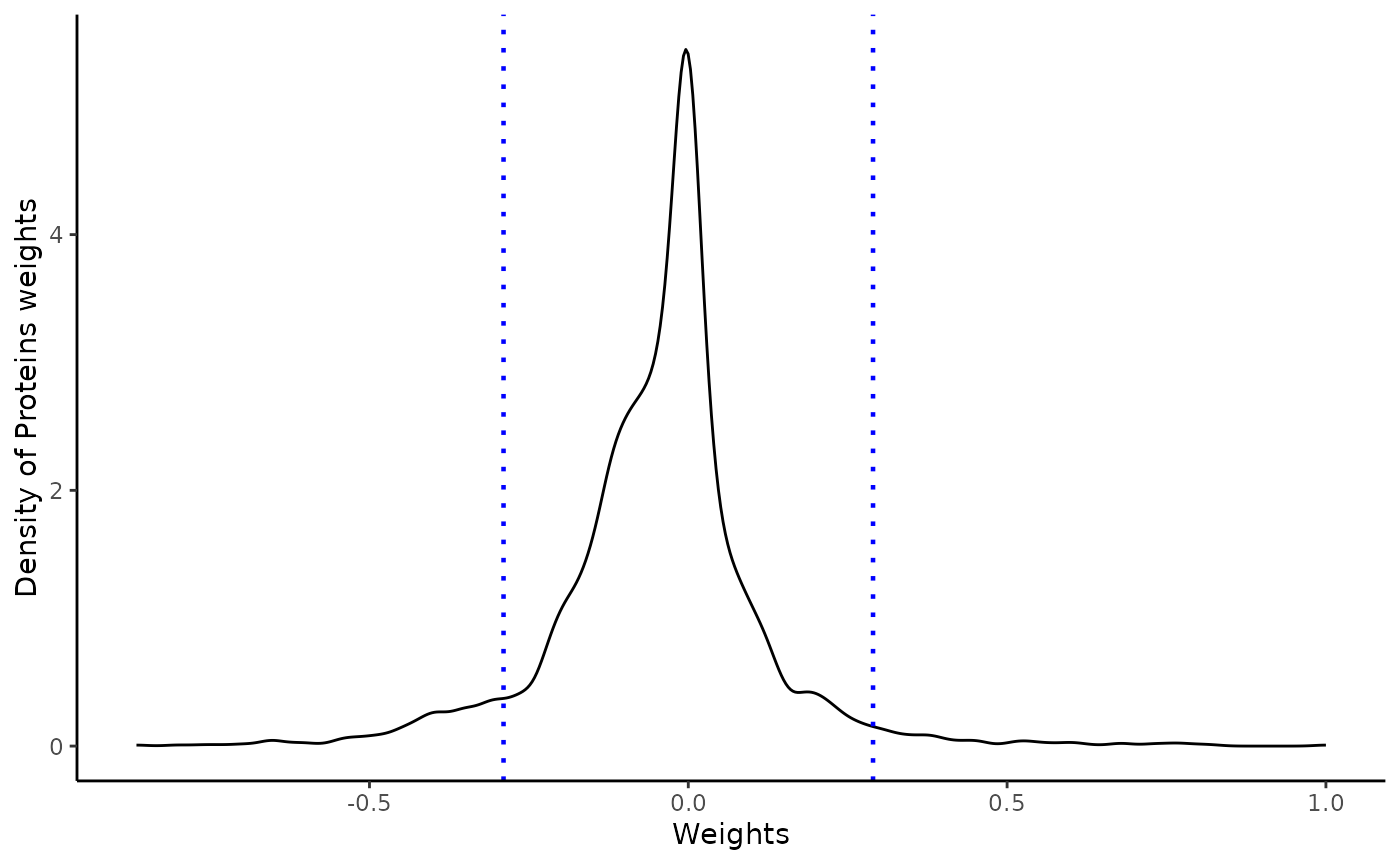
Weights vary from -1 to +1, and provide a score for how strong each feature relates to each factor, hence allowing a biological interpretation of the latent factors. Features with no association with the factor have values close to zero, while genes with strong association with the factor have large absolute values.
The sign of the weight indicates the direction of the effect:
A positive weight indicates that the feature has higher levels in the samples with positive factor values.
A negative weight indicate higher levels in samples with negative factor values.
The extract_weigthsfunction enable to extract the
weights on the desired factor at a defined absolute threshold, and
return an object with positive and negative weights above and below this
threshold respectively. It also returns QC plots
A distribution of the feature weights for each omic layer at the designated factor
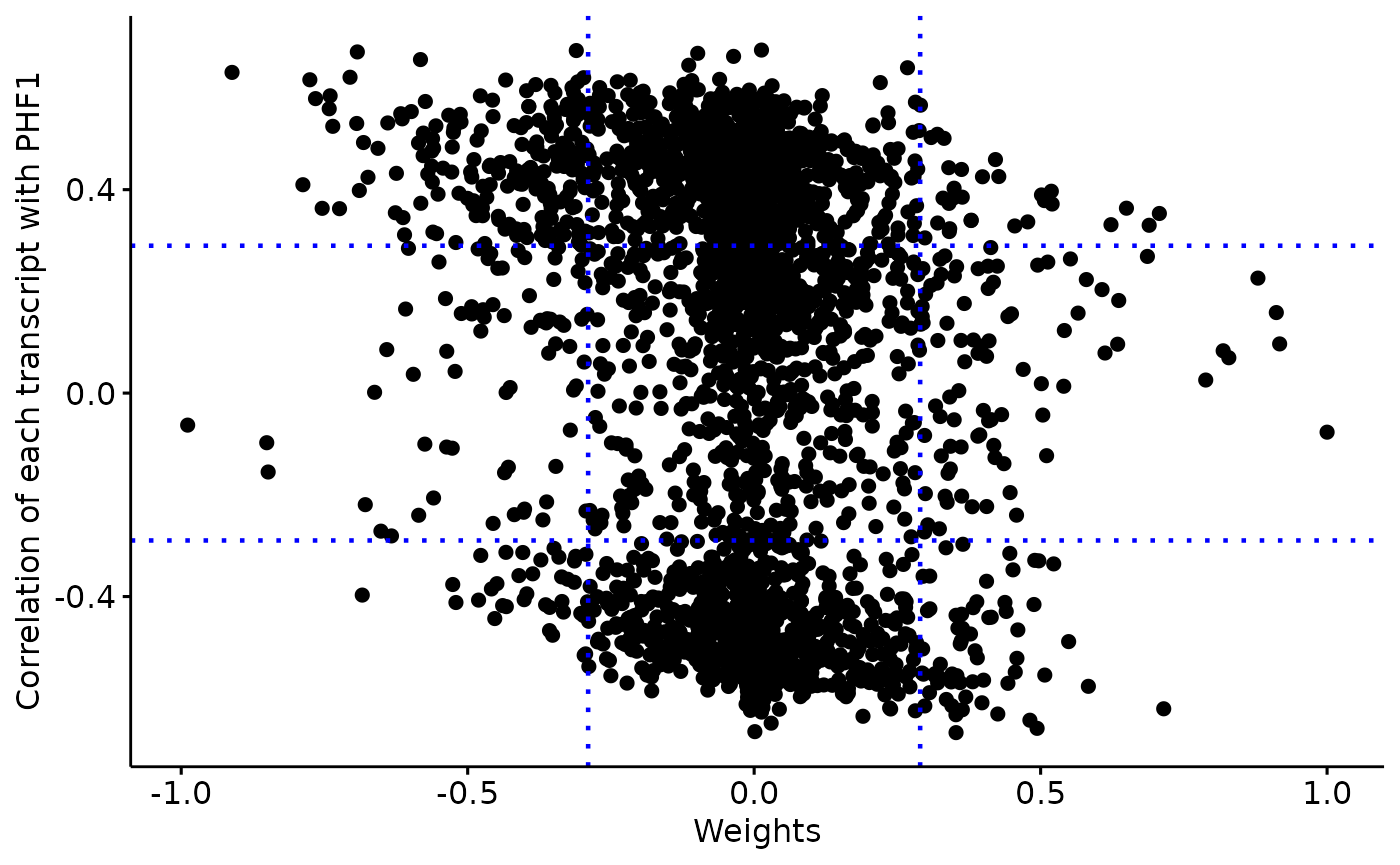
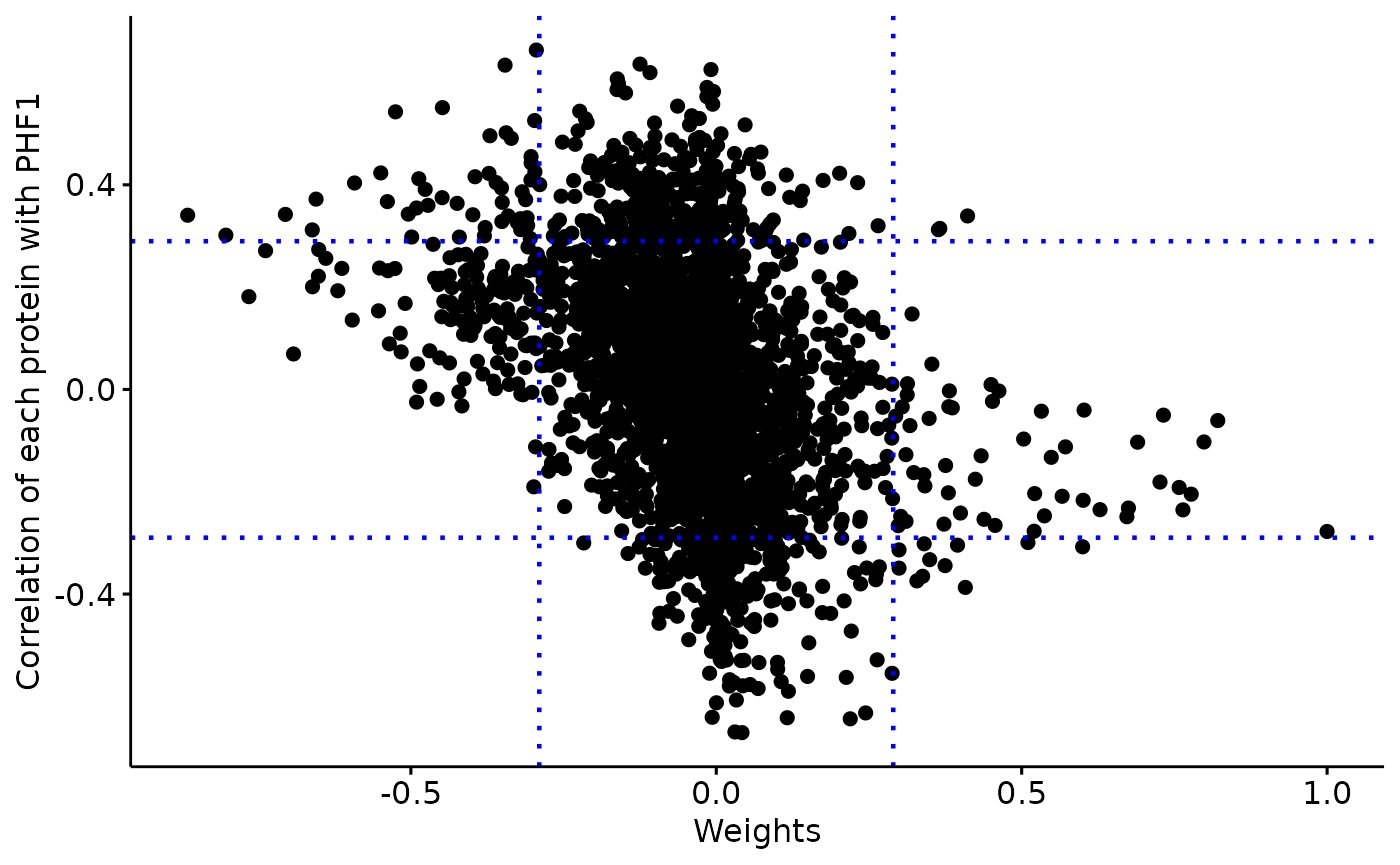
The relationship between the feature/
sense_check_variablecorrelation and weights. High weights should coincide with stronger correlation if thesense_check_variableis an important driver of variation in the designated factor.
weights=extract_weigths(integrated_object,
factor=6,
threshold=1.5,
sense_check_variable='PHF1')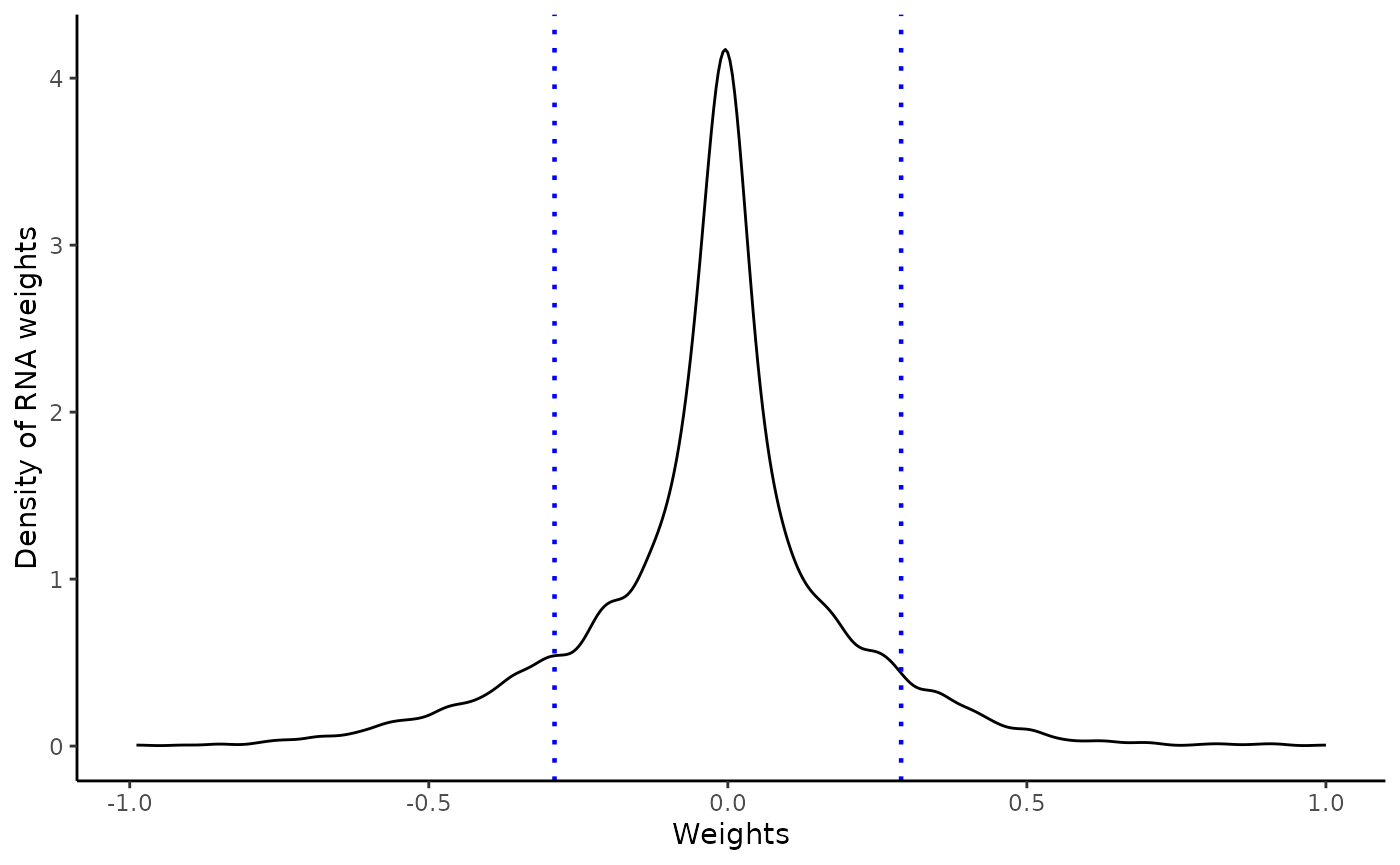
Negative weights in factor 6: Up-regulated in later stage AD
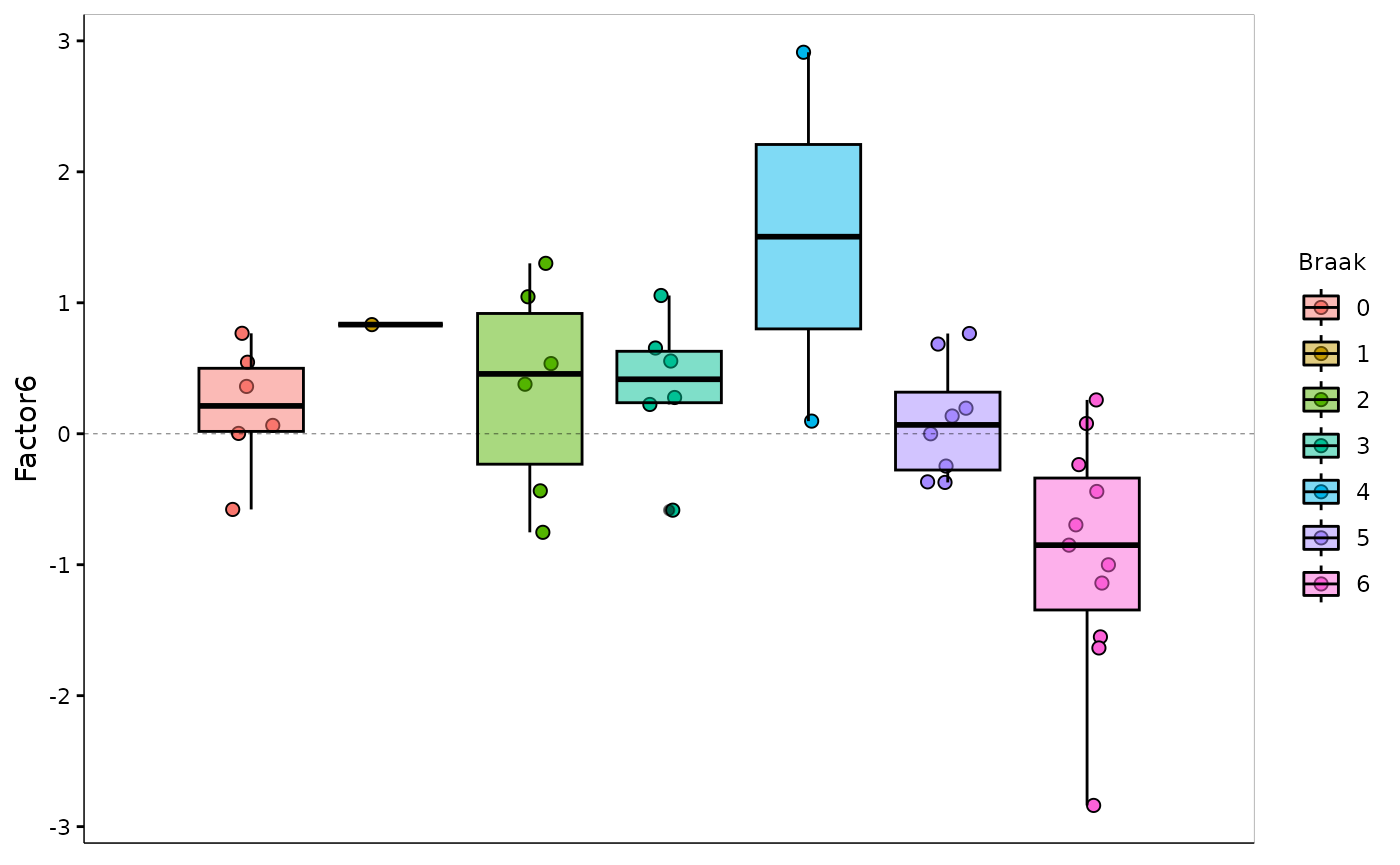
A negative weight indicates that the feature has higher levels in the samples with negative factor values, which in this analysis coincides with later Braak stages.
integrated_object@samples_metadata$Braak=as.factor(integrated_object@samples_metadata$Braak)
MOFA2::plot_factor(integrated_object,
factors = 6,
color_by = "Braak",
add_violin = TRUE,
dodge = TRUE
)
Selected features based on defined threshold
rlength(weights\(weights\)ranked_weights_negative$rna)` RNAs have weights below -0.3 in factor 6176 proteins have weights below -0.3 in factor 6
Some driving features may overlap between transcriptomics and proteomics
intersect(weights$weights$ranked_weights_negative$rna,weights$weights$ranked_weights_negative$protein)## [1] "AKAP5" "FLNA" "HSPG2" "AHNAK" "COL6A2" "CAPG" "TAGLN"
## [8] "FABP7" "F13A1" "C3" "COL14A1" "ANXA2" "C1QC" "CRABP1"
## [15] "DCN" "COL1A1" "CALB2"- 7 features overlap between omic layers
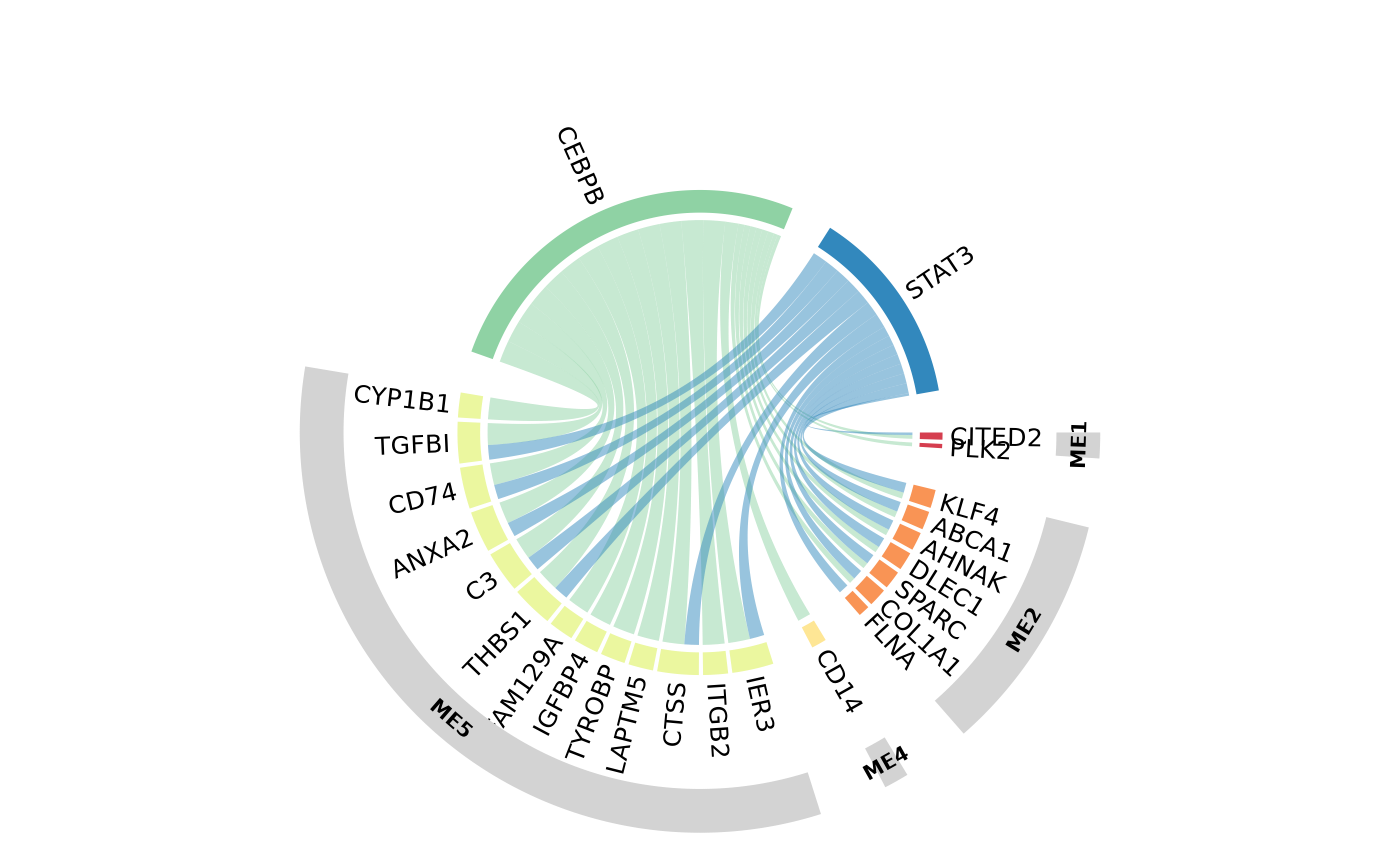
Multi-omics network
Here we choose a moderate correlation threshold to draw edges between omic features (0.4)
The same analysis can be repeated with an increased correlation threshold, which will yield more modules of strongly co-expressed features.
Weights_down_n=multiomics_network(multiassay=multiomics_object,
list=weights$weights$ranked_weights_negative,
correlation_threshold =0.4,
filter_string_50= TRUE)Community detection within multi-omic network
The next step of the analysis is to find densily co-expressed communities of proteins and RNAs within the multi-omics network.
This function tries to find densely connected subgraphs in a graph by calculating the leading non-negative eigenvector of the modularity matrix of the graph.
communities <- communities_network(igraph=Weights_down_n$graph,
community_detection='leading_eigen')
df <- purrr::map_dfr(
.x = communities$communities,
.f = ~ tibble::enframe(
x = .x,
name = NULL,
value = "feature"
),
.id = "community"
)
DT::datatable(
df,
rownames = FALSE,
extensions = 'Buttons',
options = list(
dom = 'Blfrtip',
buttons = list('colvis', list(extend = 'copy', exportOptions = list(columns=':visible')),
list(extend = 'csv', exportOptions = list(columns=':visible')),
list(extend = 'excel', exportOptions = list(columns=':visible')))))- 5 communities are detected in the multi-omics network.
- Most communities are omic dependent, since generally there is more intra omic correlation than inter omic correlation
Community 1
community_1=community_graph(igraph=Weights_down_n$graph,
community_object=communities$community_object,
community=1)
interactive_network(igraph=community_1$graph,communities=FALSE)Community 4
community_4=community_graph(igraph=Weights_down_n$graph,
community_object=communities$community_object,
community=4)
interactive_network(igraph=community_4$graph,communities=FALSE)Community hubs
- Features are more or less connected in the network, represented by their hub score:
## LY6H_protein TBC1D24_protein RAB12_protein CSNK1D_protein PMM1_protein
## 1.00000000 0.96067207 0.95018446 0.91373083 0.87930158
## PDK2_protein LAMTOR1_protein ARMT1_protein ATP6AP1_protein EIF1_protein
## 0.85923738 0.85679461 0.83362816 0.83116292 0.82305924
## USP46_protein PRUNE2_protein C2CD5_protein MPC2_protein SCG2_protein
## 0.82070521 0.81406855 0.80837977 0.79741826 0.79489801
## GMFG_protein AP1S1_protein SYNPR_protein APEH_protein COL1A1_protein
## 0.78972525 0.78775116 0.77560088 0.76911869 0.73923502
## PRKACA_protein POR_protein CPE_protein TTC7B_protein HLA-A_protein
## 0.72856261 0.72831672 0.71370465 0.70863268 0.69038092
## SNRNP70_protein SNX32_protein CAB39L_protein SYP_protein GNG5_protein
## 0.67340658 0.64531133 0.64488836 0.63591664 0.62045081
## ALDH3A2_protein SCN3B_protein EXOC5_protein SRP54_protein MAST1_protein
## 0.60033147 0.58197165 0.57721314 0.57670350 0.56005774
## ARMC8_protein COPG1_protein WIPI2_protein TUBG1_protein GABRA1_protein
## 0.54501347 0.53781567 0.53503646 0.52329867 0.51294443
## NECTIN1_protein ITPR1_protein SNRPA1_protein GUCY1A2_protein DLGAP3_protein
## 0.45166801 0.44363032 0.41212481 0.39655465 0.36301002
## C1QB_rna ATAD3B_protein CYBB_rna F13A1_rna CCR1_rna
## 0.26034395 0.24893281 0.22441530 0.17817574 0.13574288
## CD14_rna CD53_rna CX3CR1_rna LPCAT2_rna LIPG_rna
## 0.12660867 0.11629988 0.10989585 0.08313190 0.05830927
## PAX8_rna
## 0.02647772- Top gene in module 4 is LY6H_protein
- TREM2 or CLU, known AD risk genes expressed on microglia are present here
- From prior knowledge, many of the genes in module 4 are related to microglial activation
Cell type enrichment of modules
Here we proceed with a EWCE analysis to check if modules
are enriched in specific cell types.
cell_type <- cell_type_enrichment(
multiassay =multiomics_object,
communities = lapply(communities$communities,function(x){sub("\\_.*", "",x )}),
ctd= ctd
)
cell_type$plots## $`1`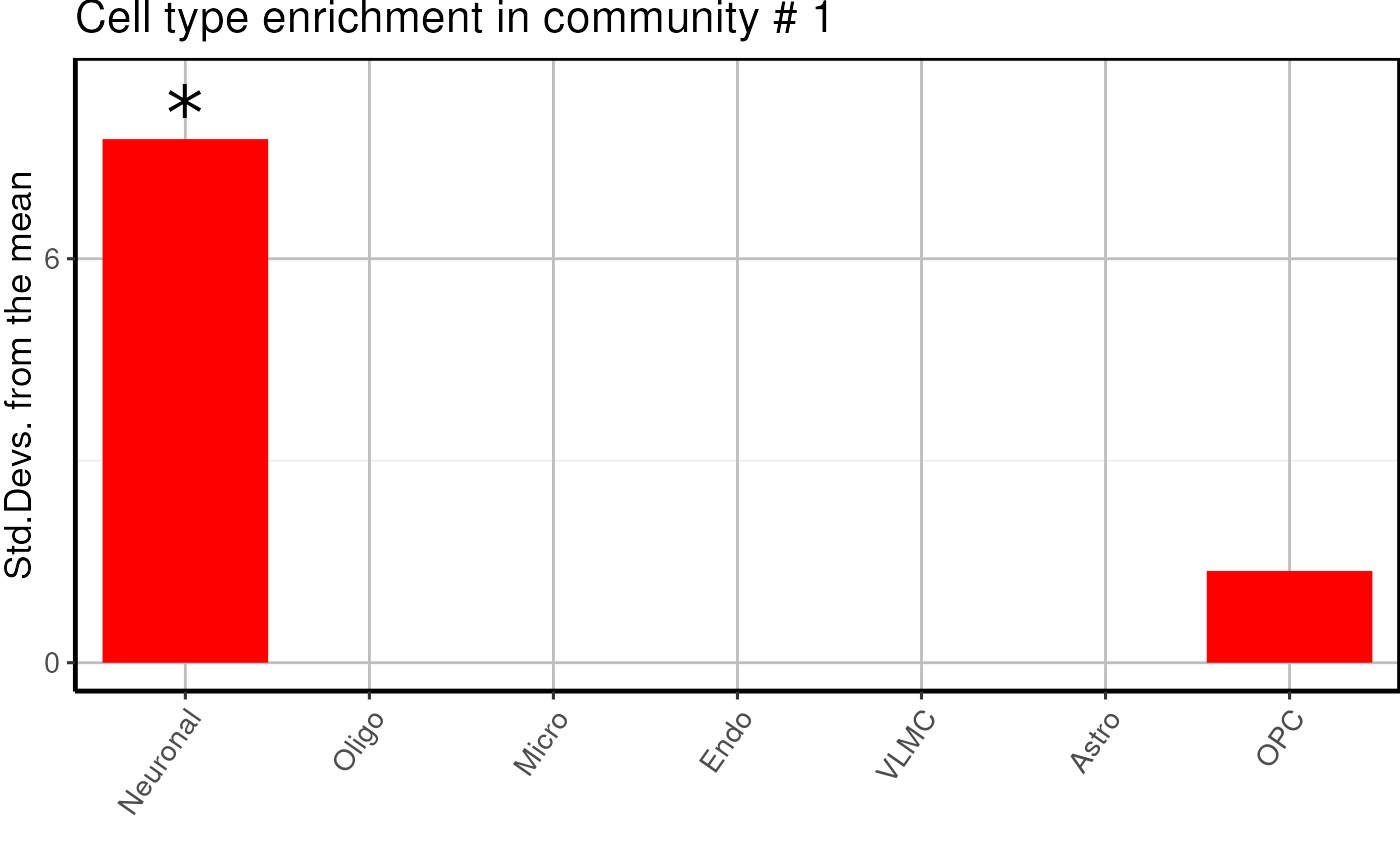
##
## $`2`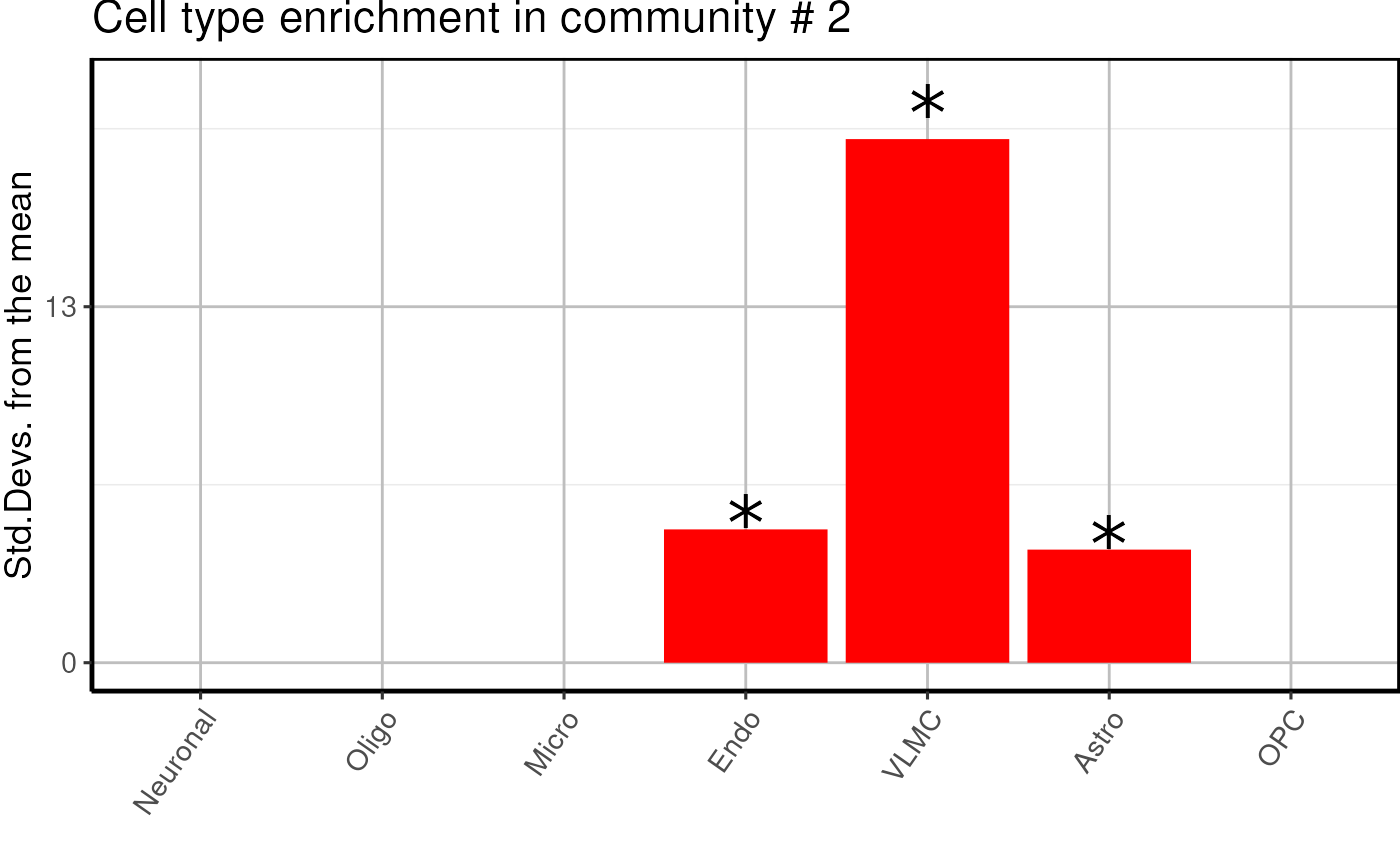
##
## $`3`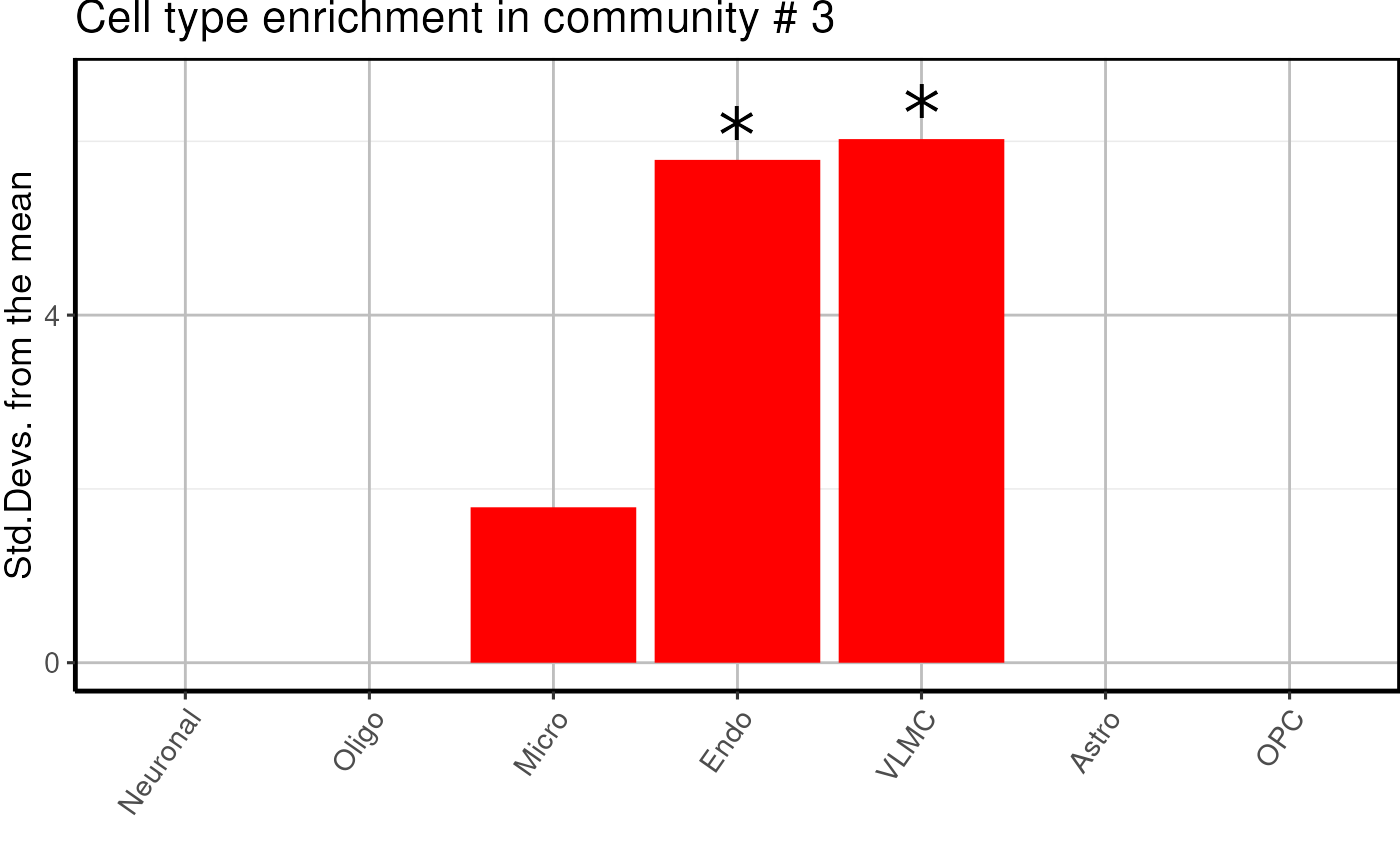
##
## $`4`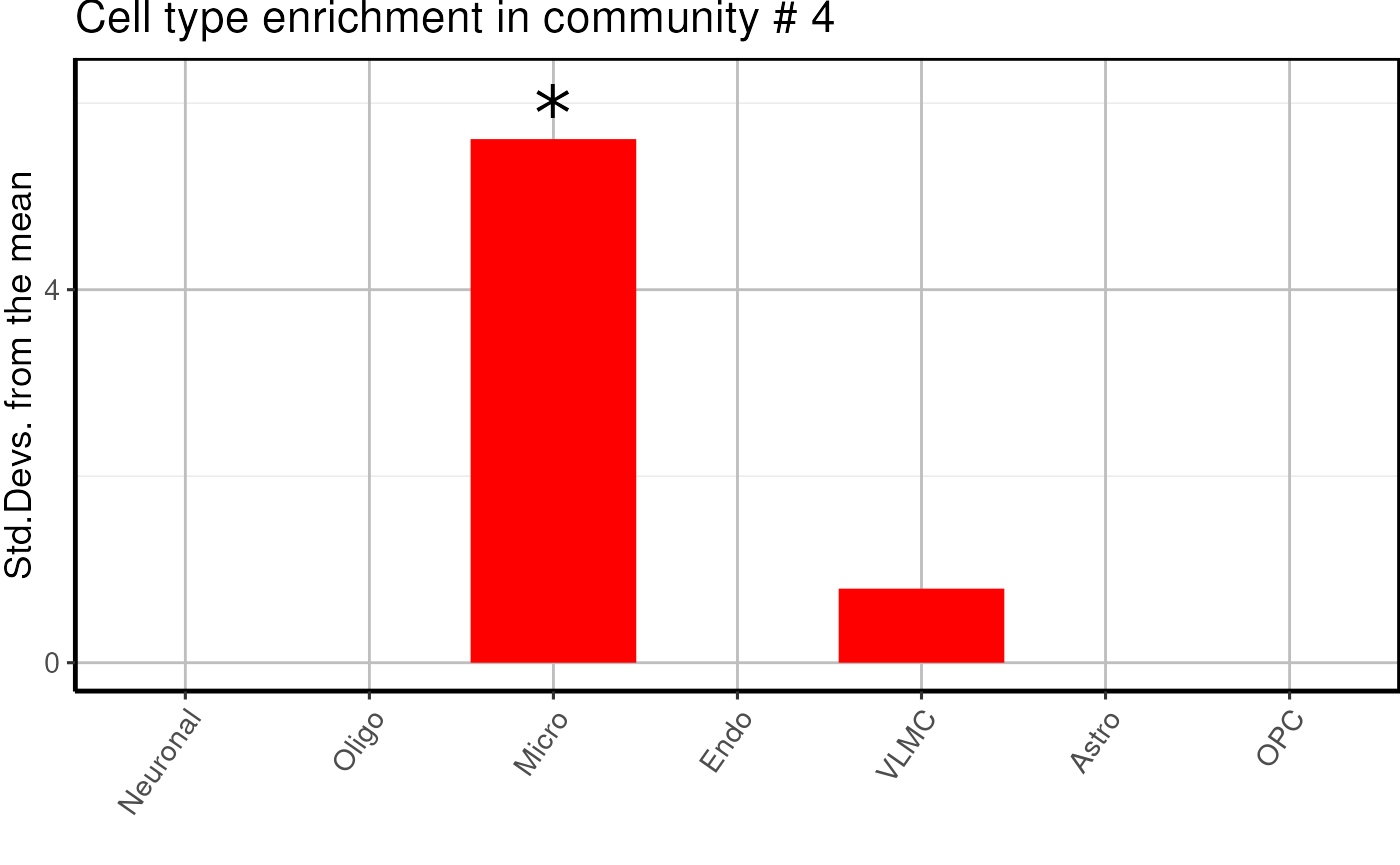
##
## $`5`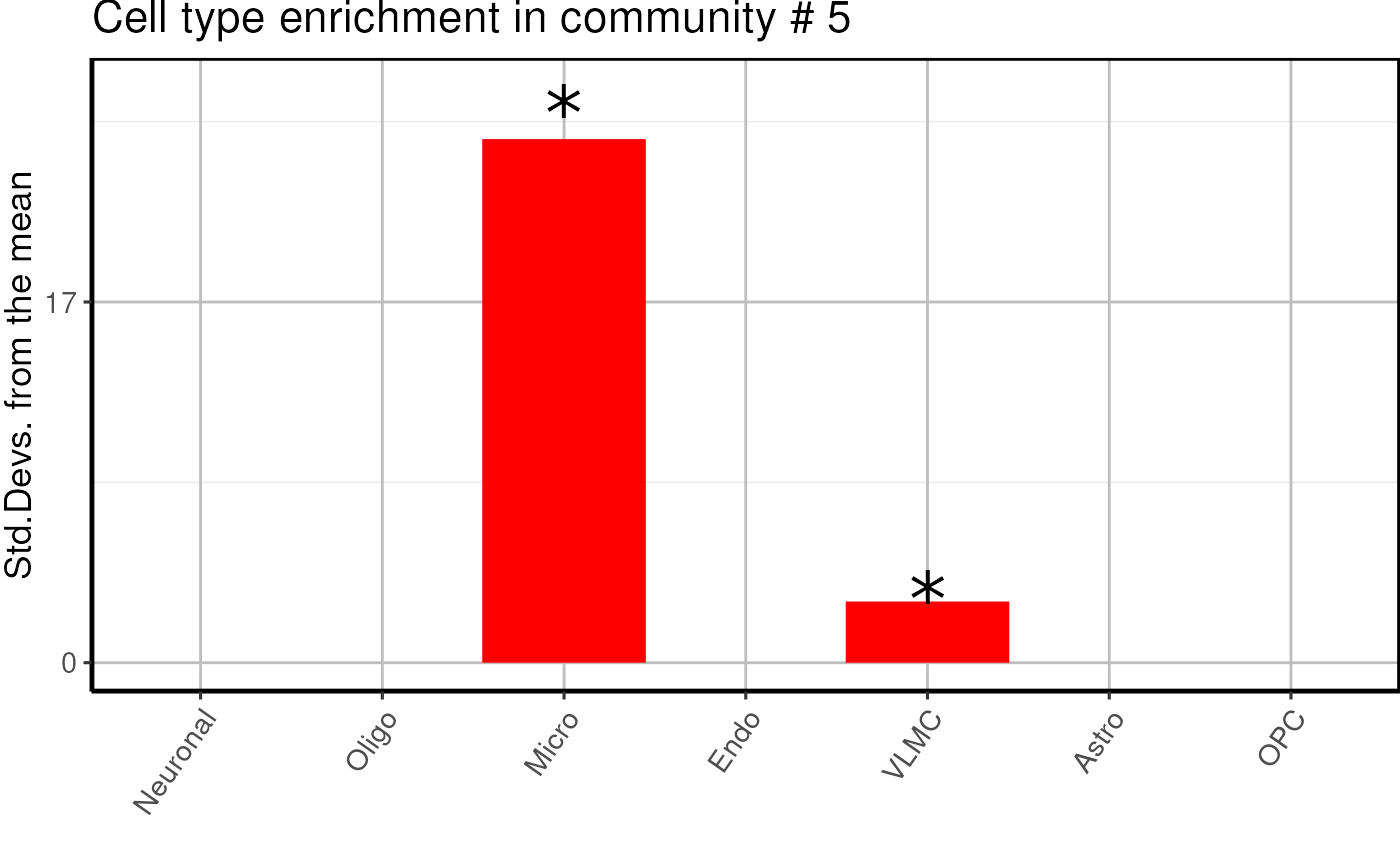
- We find one neuronal, one microglial and two undefined glial enriched modules.
Pathway enrichment analysis
communities_l = lapply(communities$communities,function(x){sub("\\_.*", "",x )})
enrichment1=pathway_analysis_enrichr(communities_l$`1`,plot=5)## Welcome to enrichR
## Checking connection ...## Enrichr ... Connection is Live!
## FlyEnrichr ... Connection is available!
## WormEnrichr ... Connection is available!
## YeastEnrichr ... Connection is available!
## FishEnrichr ... Connection is available!## Uploading data to Enrichr... Done.
## Querying GO_Molecular_Function_2021... Done.
## Querying GO_Cellular_Component_2021... Done.
## Querying GO_Biological_Process_2021... Done.
## Querying WikiPathways_2021_Human... Done.
## Querying Reactome_2016... Done.
## Querying KEGG_2021_Human... Done.
## Querying MSigDB_Hallmark_2020... Done.
## Querying BioCarta_2016... Done.
## Parsing results... Done.## ℹ Output is returned as a list!
enrichment2=pathway_analysis_enrichr(communities_l$`2`,plot=5)## Welcome to enrichR
## Checking connection ...
## Enrichr ... Connection is Live!
## FlyEnrichr ... Connection is available!
## WormEnrichr ... Connection is available!
## YeastEnrichr ... Connection is available!
## FishEnrichr ... Connection is available!## Uploading data to Enrichr... Done.
## Querying GO_Molecular_Function_2021... Done.
## Querying GO_Cellular_Component_2021... Done.
## Querying GO_Biological_Process_2021... Done.
## Querying WikiPathways_2021_Human... Done.
## Querying Reactome_2016... Done.
## Querying KEGG_2021_Human... Done.
## Querying MSigDB_Hallmark_2020... Done.
## Querying BioCarta_2016... Done.
## Parsing results... Done.## ℹ Output is returned as a list!
enrichment3=pathway_analysis_enrichr(communities_l$`3`,plot=5)## Welcome to enrichR
## Checking connection ...
## Enrichr ... Connection is Live!
## FlyEnrichr ... Connection is available!
## WormEnrichr ... Connection is available!
## YeastEnrichr ... Connection is available!
## FishEnrichr ... Connection is available!## Uploading data to Enrichr... Done.
## Querying GO_Molecular_Function_2021... Done.
## Querying GO_Cellular_Component_2021... Done.
## Querying GO_Biological_Process_2021... Done.
## Querying WikiPathways_2021_Human... Done.
## Querying Reactome_2016... Done.
## Querying KEGG_2021_Human... Done.
## Querying MSigDB_Hallmark_2020... Done.
## Querying BioCarta_2016... Done.
## Parsing results... Done.## ℹ Output is returned as a list!
enrichment4=pathway_analysis_enrichr(communities_l$`4`,plot=5)## Welcome to enrichR
## Checking connection ...
## Enrichr ... Connection is Live!
## FlyEnrichr ... Connection is available!
## WormEnrichr ... Connection is available!
## YeastEnrichr ... Connection is available!
## FishEnrichr ... Connection is available!## Uploading data to Enrichr... Done.
## Querying GO_Molecular_Function_2021... Done.
## Querying GO_Cellular_Component_2021... Done.
## Querying GO_Biological_Process_2021... Done.
## Querying WikiPathways_2021_Human... Done.
## Querying Reactome_2016... Done.
## Querying KEGG_2021_Human... Done.
## Querying MSigDB_Hallmark_2020... Done.
## Querying BioCarta_2016... Done.
## Parsing results... Done.## ℹ Output is returned as a list!
enrichment5=pathway_analysis_enrichr(communities_l$`5`,plot=5)## Welcome to enrichR
## Checking connection ...
## Enrichr ... Connection is Live!
## FlyEnrichr ... Connection is available!
## WormEnrichr ... Connection is available!
## YeastEnrichr ... Connection is available!
## FishEnrichr ... Connection is available!## Uploading data to Enrichr... Done.
## Querying GO_Molecular_Function_2021... Done.
## Querying GO_Cellular_Component_2021... Done.
## Querying GO_Biological_Process_2021... Done.
## Querying WikiPathways_2021_Human... Done.
## Querying Reactome_2016... Done.
## Querying KEGG_2021_Human... Done.
## Querying MSigDB_Hallmark_2020... Done.
## Querying BioCarta_2016... Done.
## Parsing results... Done.## ℹ Output is returned as a list!
enrichment1$plot$Reactome_2016+theme(axis.text=element_text(size=12),
axis.title=element_text(size=12),
strip.text = element_text(size = 0, margin = margin()),
legend.title = element_text(size=9))
enrichment2$plot$Reactome_2016+theme(axis.text=element_text(size=12),
axis.title=element_text(size=12),
strip.text = element_text(size = 0, margin = margin()),
legend.title = element_text(size=9))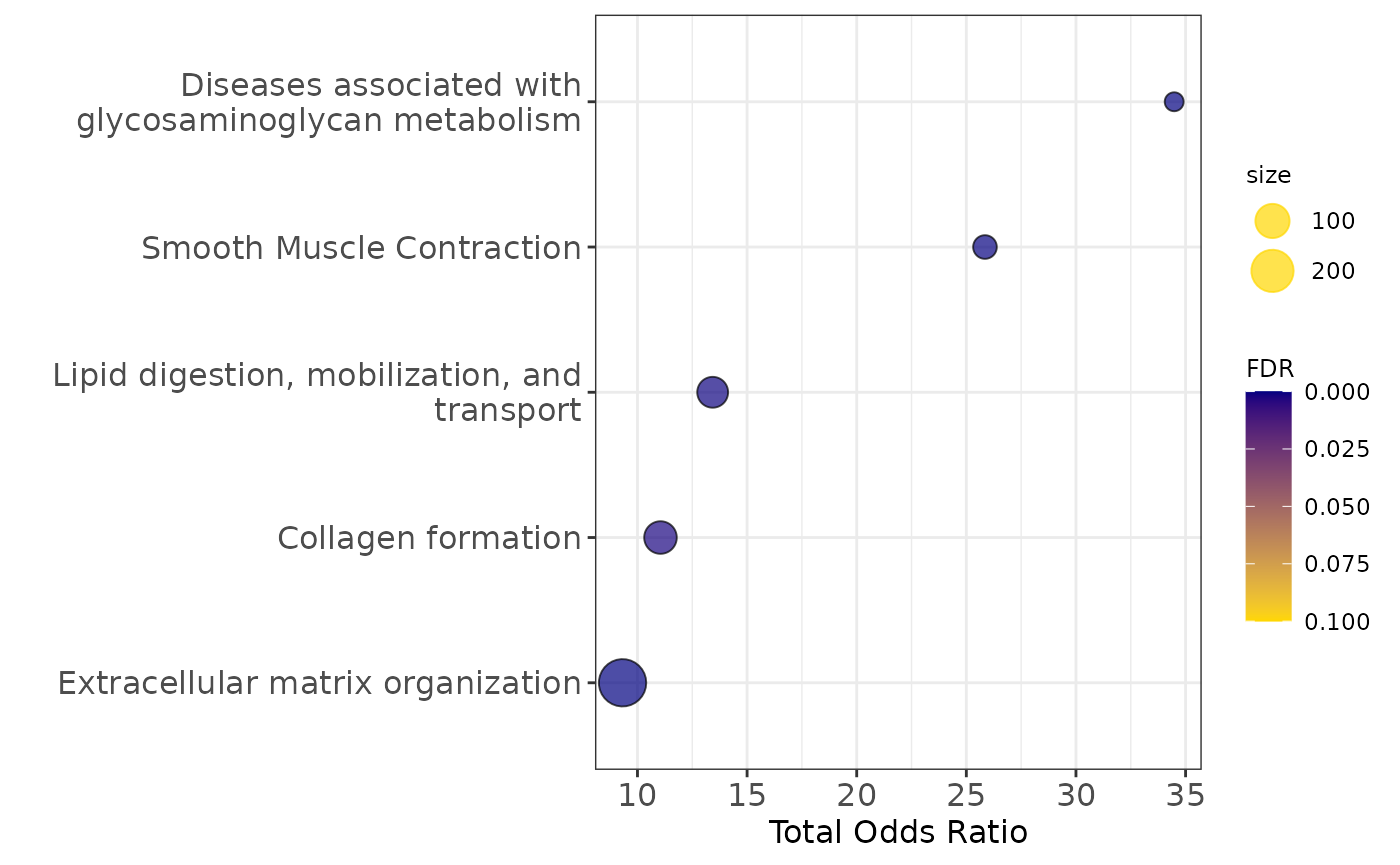
enrichment3$plot$Reactome_2016+theme(axis.text=element_text(size=12),
axis.title=element_text(size=12),
strip.text = element_text(size = 0, margin = margin()),
legend.title = element_text(size=9))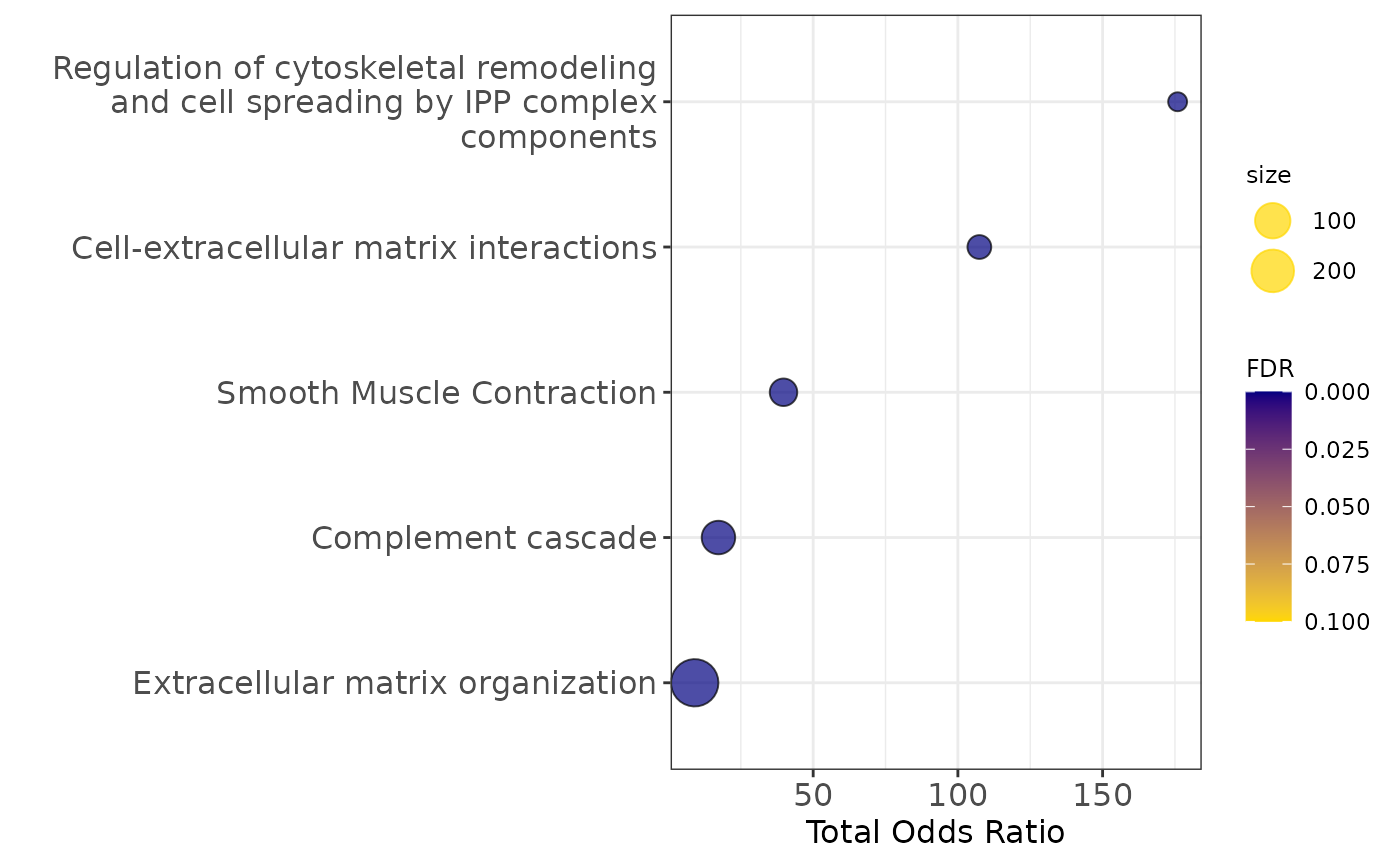
enrichment4$plot$Reactome_2016+theme(axis.text=element_text(size=12),
axis.title=element_text(size=12),
strip.text = element_text(size = 0, margin = margin()),
legend.title = element_text(size=9))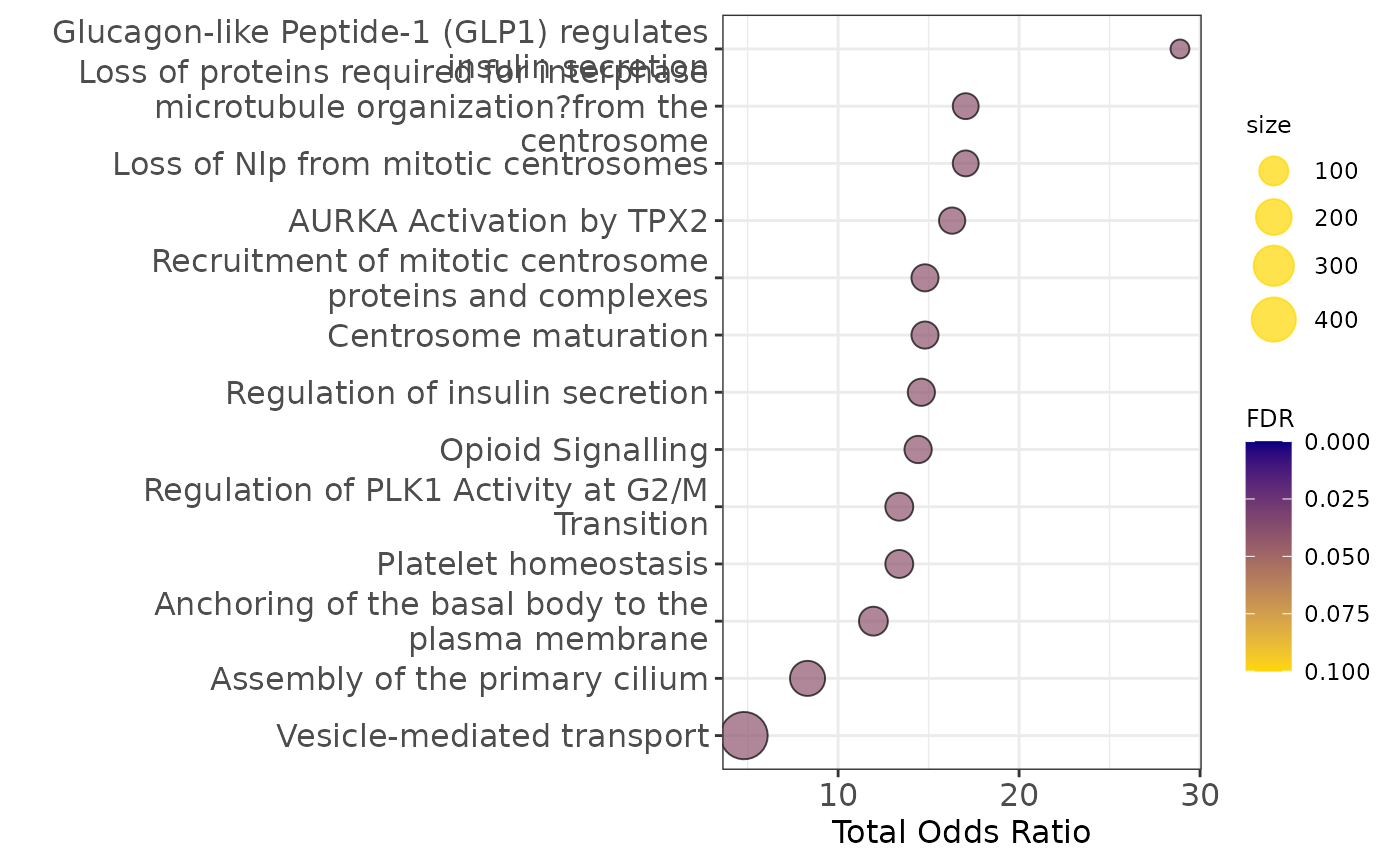
enrichment5$plot$Reactome_2016+theme(axis.text=element_text(size=12),
axis.title=element_text(size=12),
strip.text = element_text(size = 0, margin = margin()),
legend.title = element_text(size=9))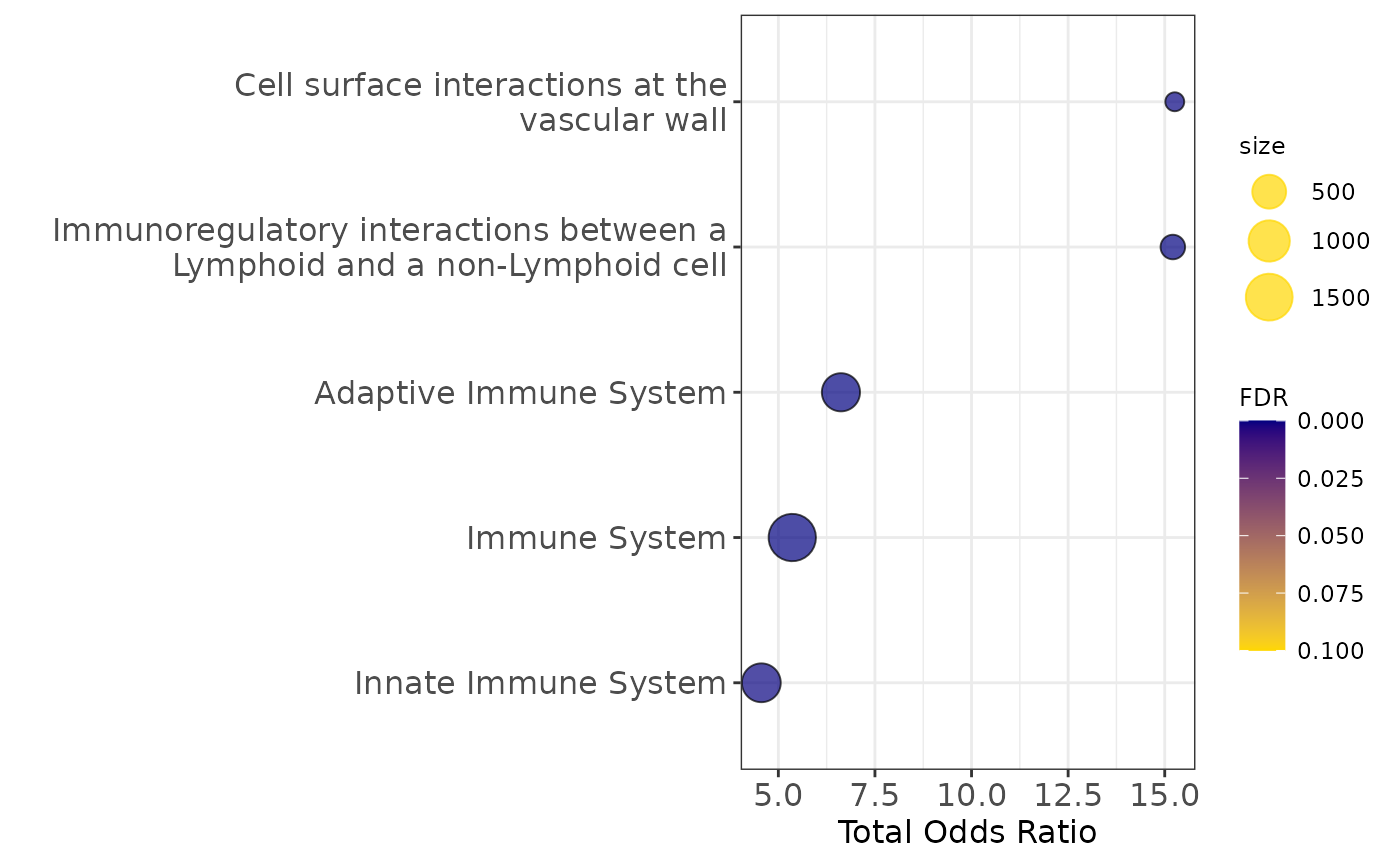
Modules correlation to neuropathology
multiomics_modules compute the module eigen value (PC1
of scaled transcriptomics/proteomics expression) and correlates it to
chosen covariates
modules=multiomics_modules(multiassay=multiomics_object,
metadata=MOFA2::samples_metadata(integrated_object),
covariates=c('inferred_pseudotime','PHF1','amyloid','pTau'),
communities=communities$communities,
filter_string_50=TRUE)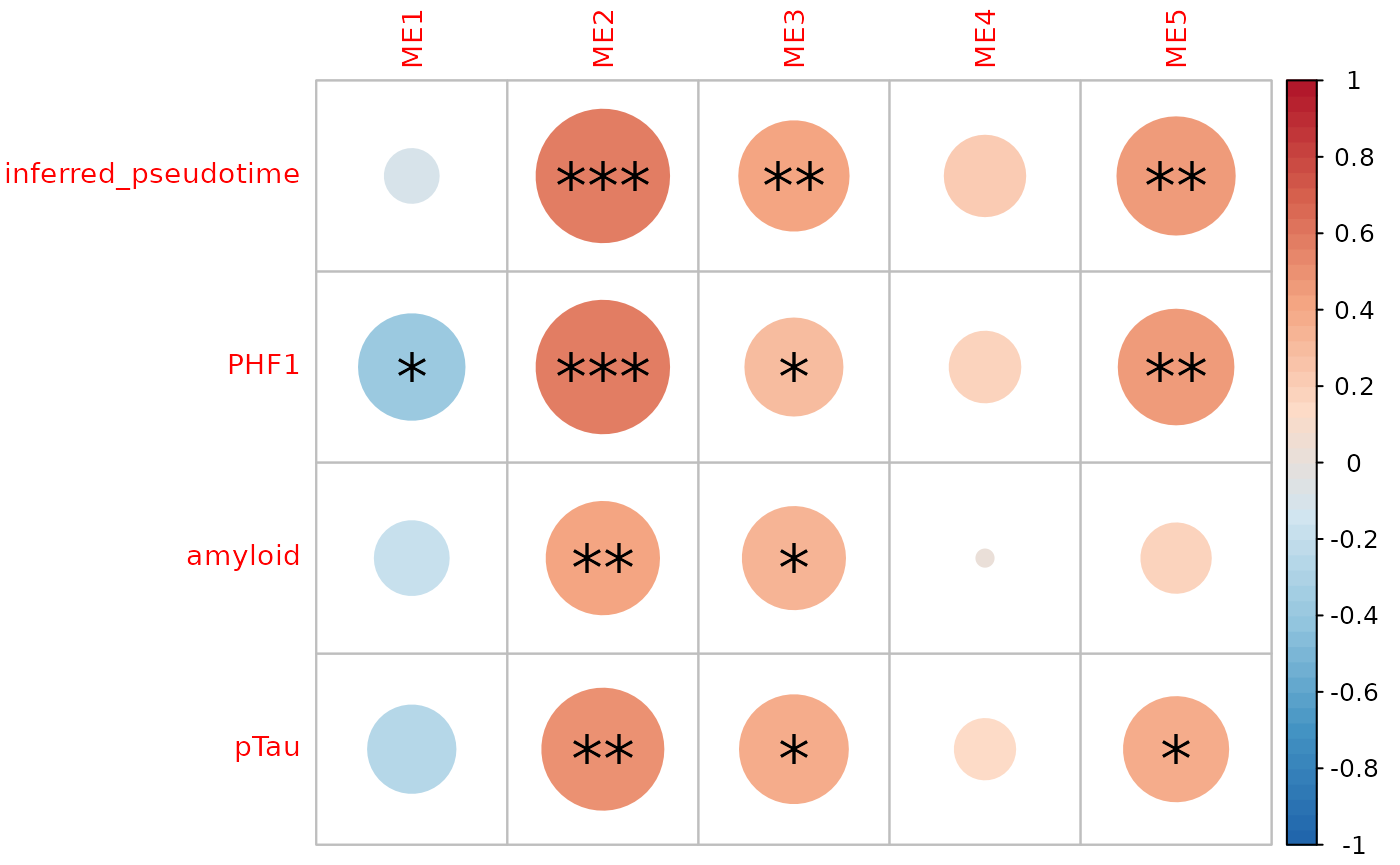
Transcription Factor - target gene enrichment
TF=Transcription_Factor_enrichment(communities=communities$communities,
weights=weights,
TF_gmt=TF_fp,
threshold = 0.2,
direction = "negative")
GeneOverlap::drawHeatmap(TF,what = c("odds.ratio"), log.scale = T, adj.p = F, cutoff = .05,
ncolused = 7, grid.col = c("Blues"), note.col = "red")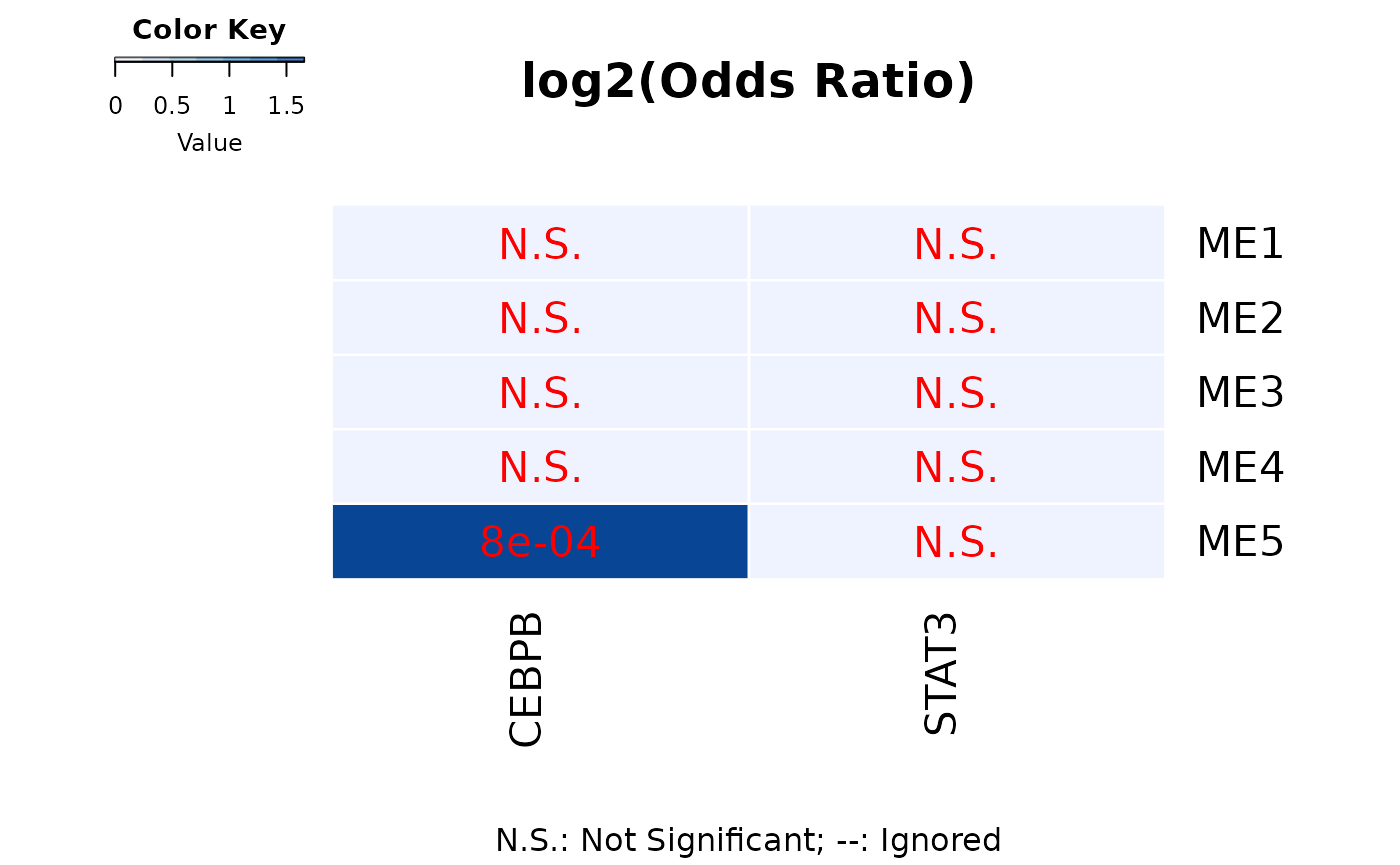
Pseudotime ordering of multi-omics modules
- Here we related each module eigen value to the pseudotime inferred from MOFA integration emmbeddings (Factors 6/8)
plot_module_trajectory(modules=modules,
covariates=c('amyloid','pTau','PHF1'),
plot_modules=c("ME1","ME2","ME3","ME4"))## `geom_smooth()` using formula = 'y ~ x'
## `geom_smooth()` using formula = 'y ~ x'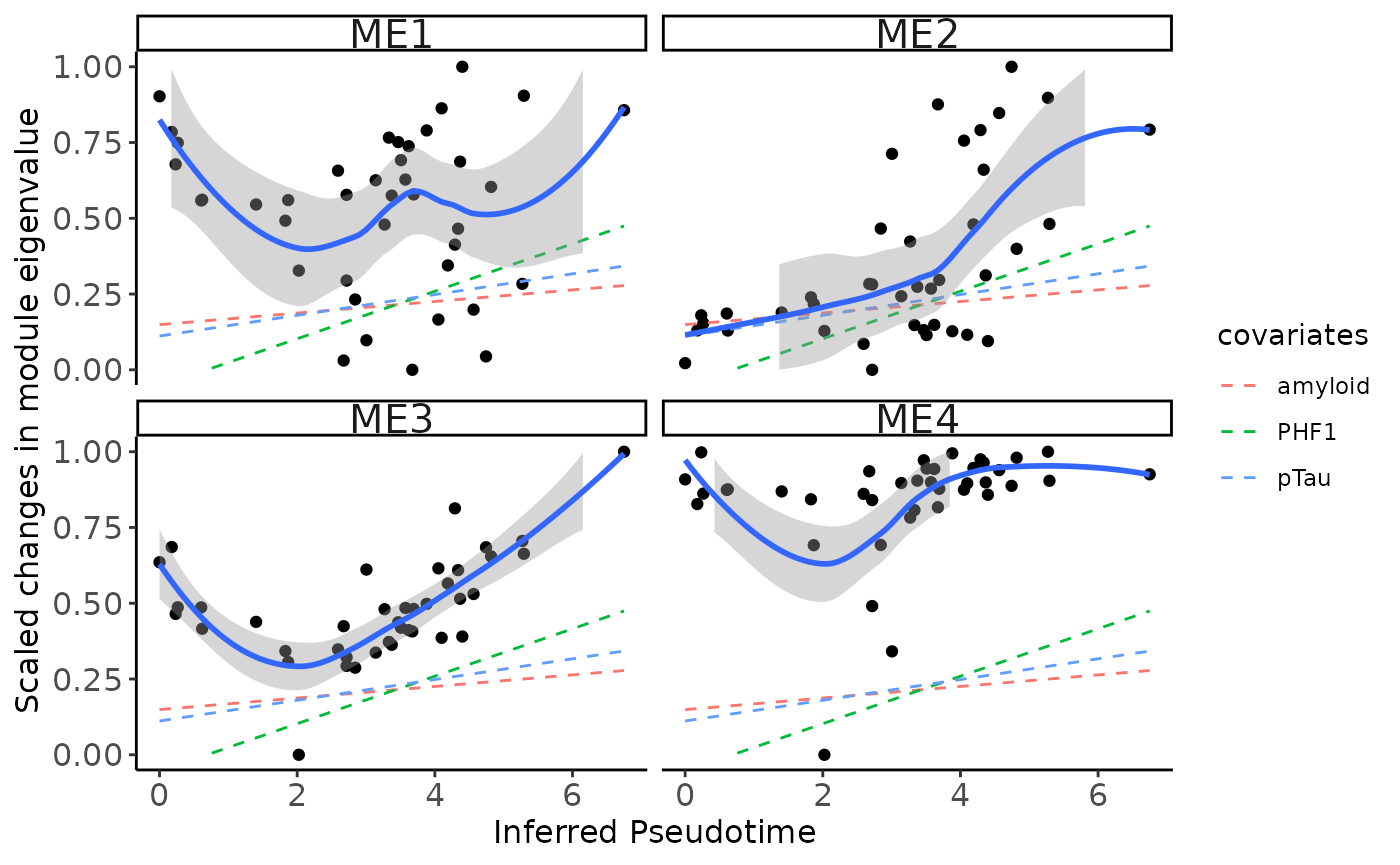
Pseudotime vs Braak
We find a stronge correlation between inferred pseudotime and PHF1, indicating that the pseudotemporal multiomics integration provides an accurate assessment of AD progression, encompassing all neuropathological measures
PHF1
modules$metadata_modules$Braak=factor(modules$metadata_modules$Braak, levels = c("0","1","2","3","4","5","6"))
modules$metadata_modules$Braak_num=as.numeric(modules$metadata_modules$Braak)
gg=ggpubr::ggscatter(modules$metadata_modules, y = "PHF1", x = "Braak_num", palette = "jco",
add = "reg.line", conf.int = TRUE, label.x = 4, add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.sep = "\n"))
gb=ggpubr::ggscatter(modules$metadata_modules, y = "PHF1", x = "inferred_pseudotime", palette = "jco",
add = "reg.line", conf.int = TRUE, add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.x = 4, label.sep = "\n"))
plot(gg)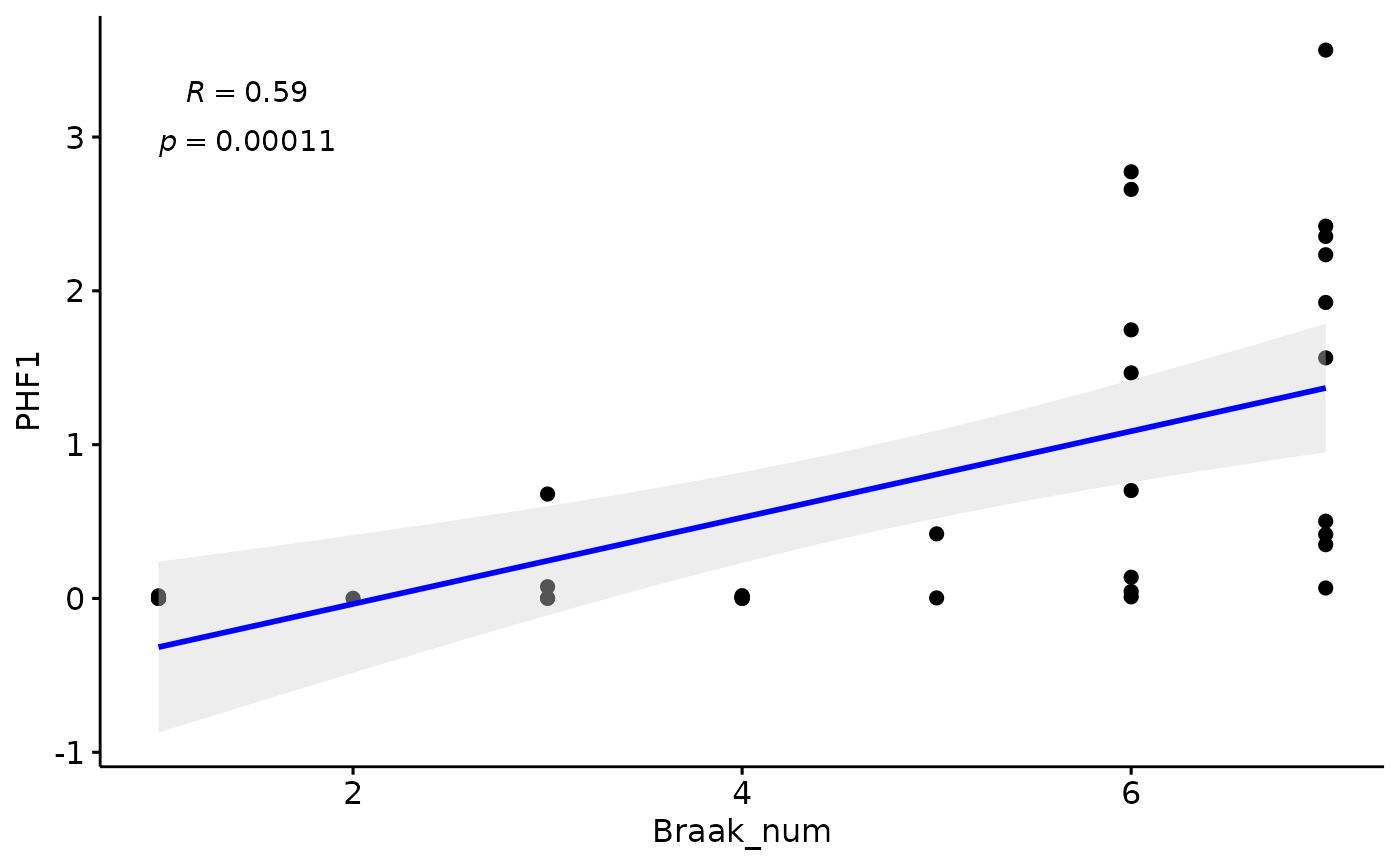
plot(gb)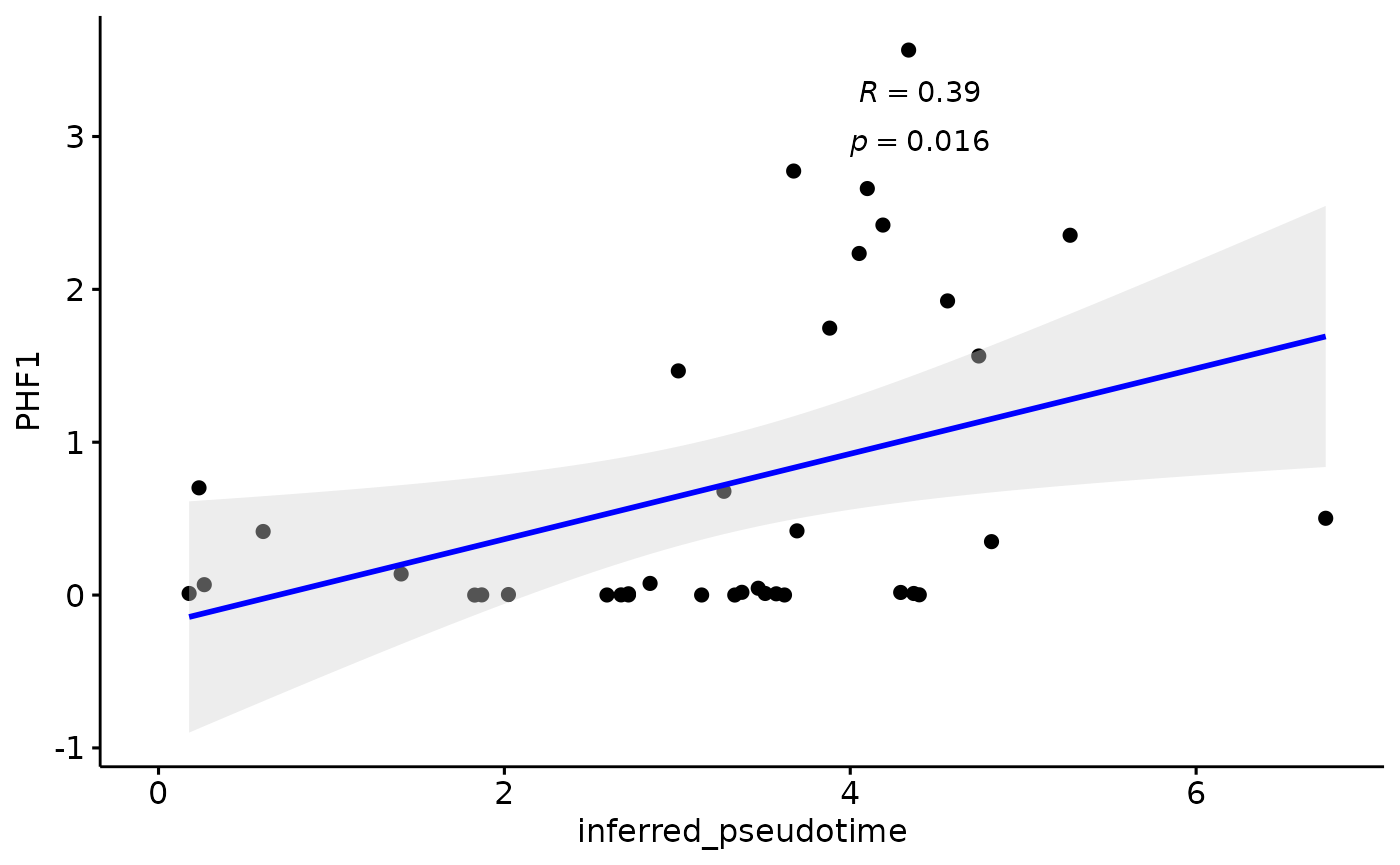
Amyloid
modules$metadata_modules$Braak=factor(modules$metadata_modules$Braak,
levels = c("0","1","2","3","4","5","6"))
modules$metadata_modules$Braak_num=as.numeric(modules$metadata_modules$Braak)
gg=ggpubr::ggscatter(modules$metadata_modules, y = "amyloid", x = "Braak_num", palette = "jco",
add = "reg.line", conf.int = TRUE, label.x = 4, add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.sep = "\n"))
gb=ggpubr::ggscatter(modules$metadata_modules, y = "amyloid", x = "inferred_pseudotime", palette = "jco",
add = "reg.line", conf.int = TRUE, add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.x = 4, label.sep = "\n"))
plot(gg)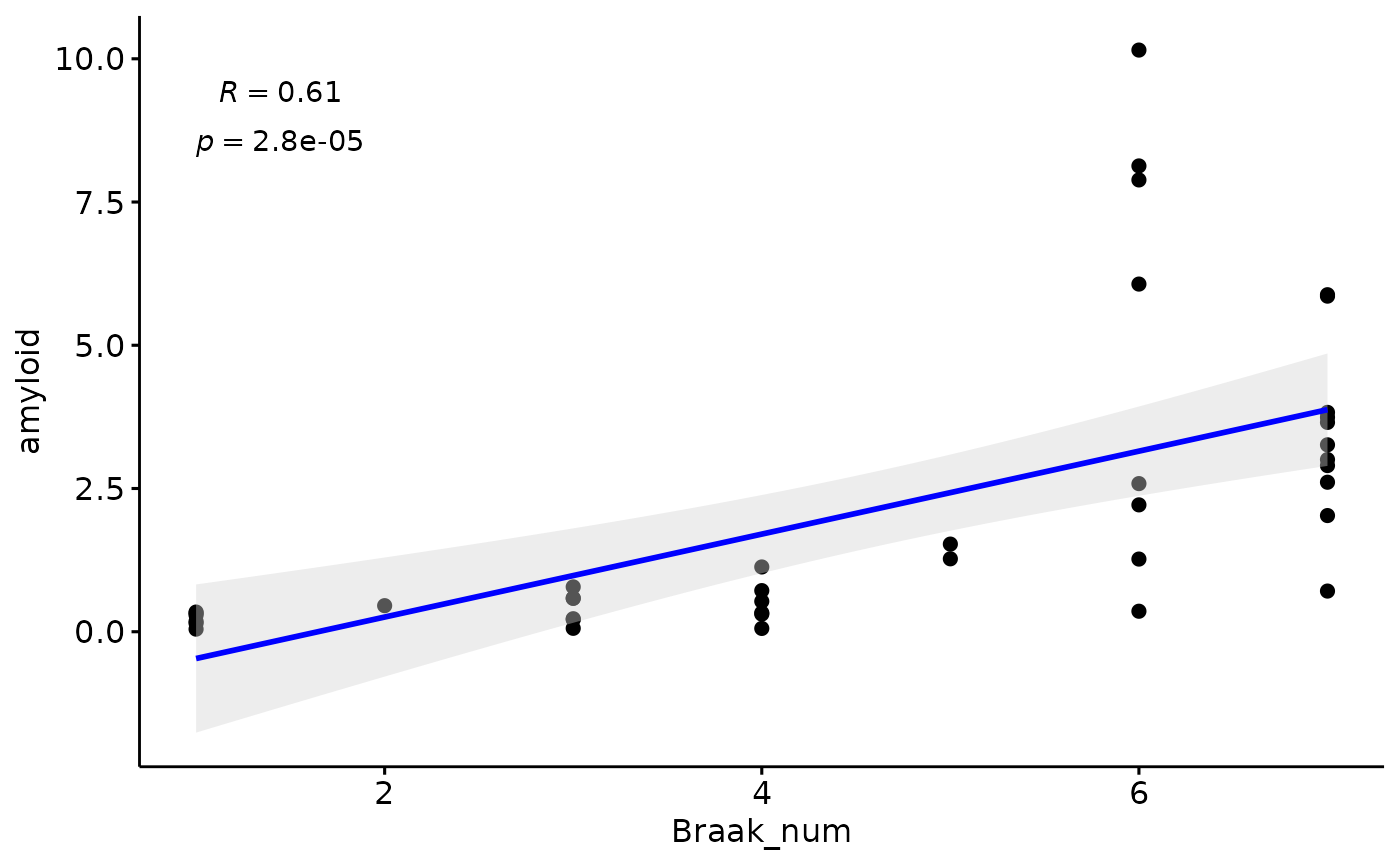
plot(gb)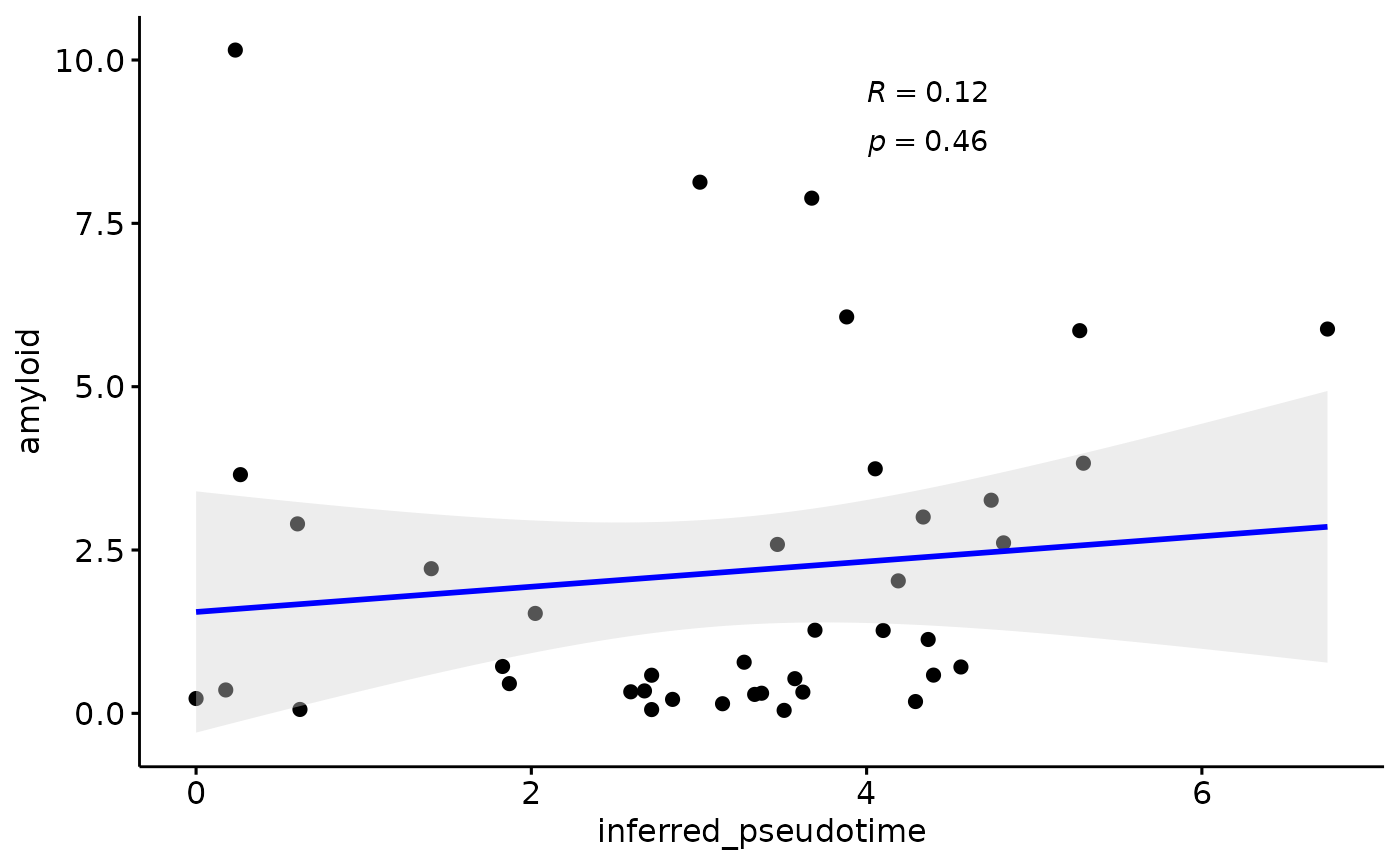
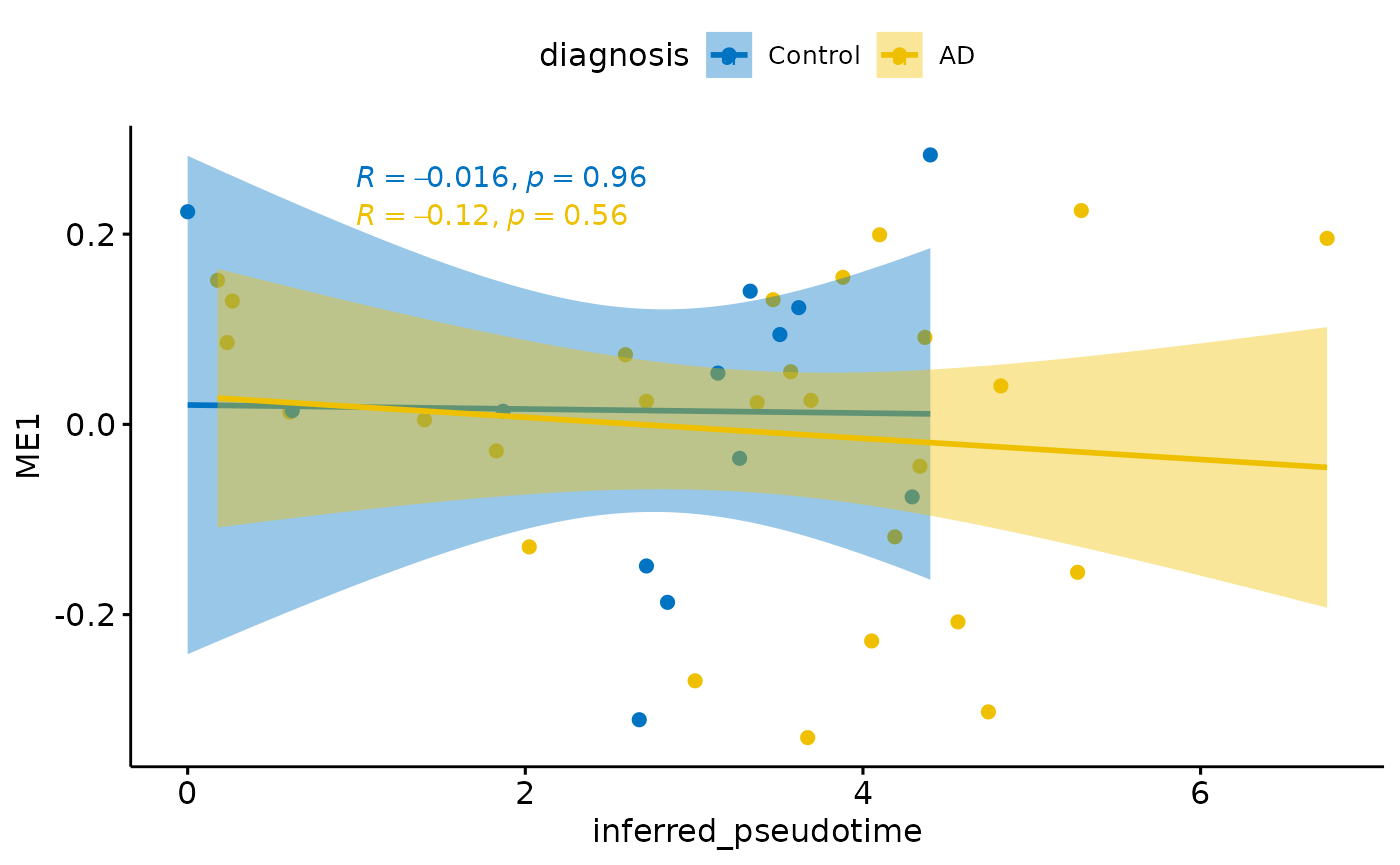
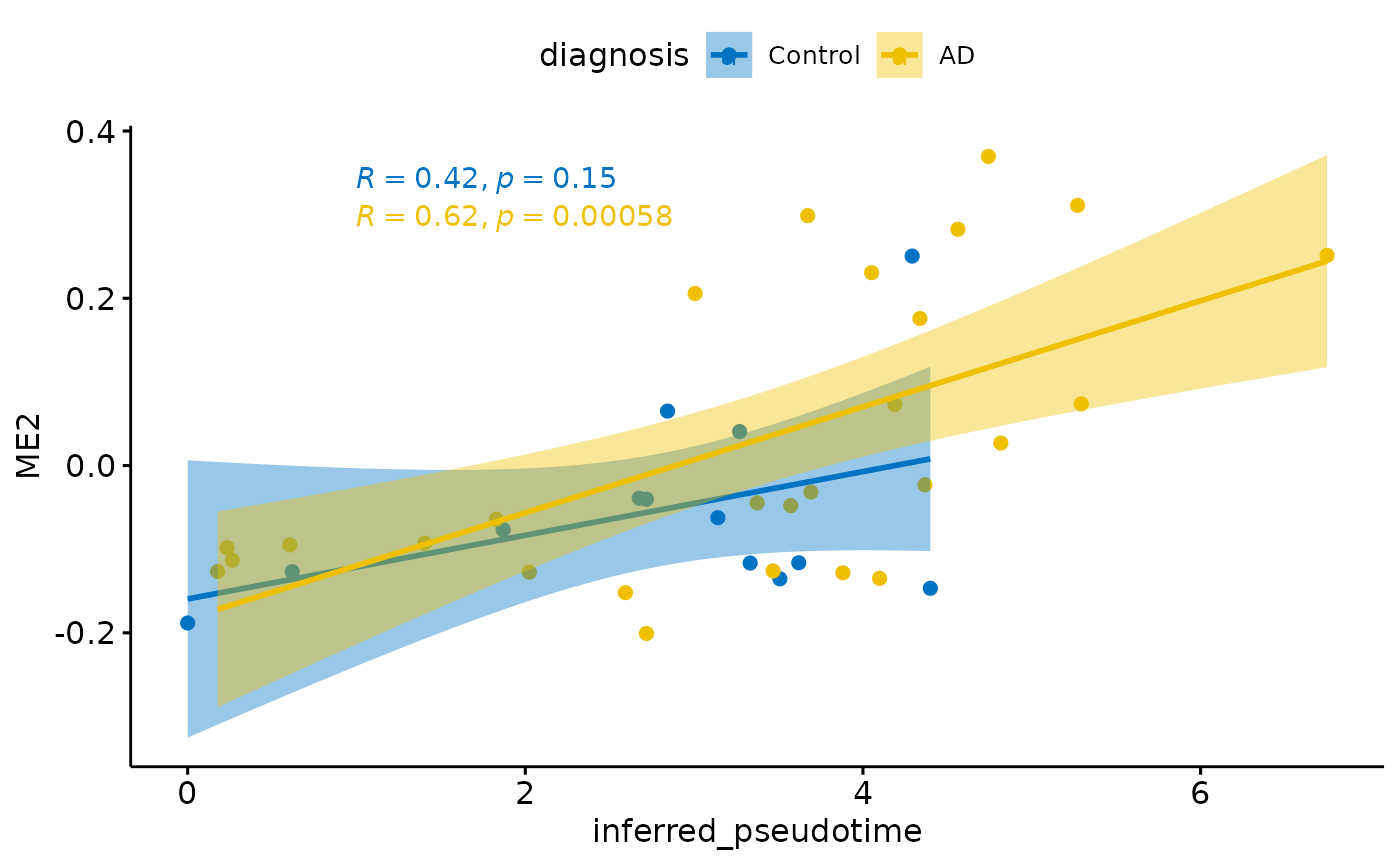
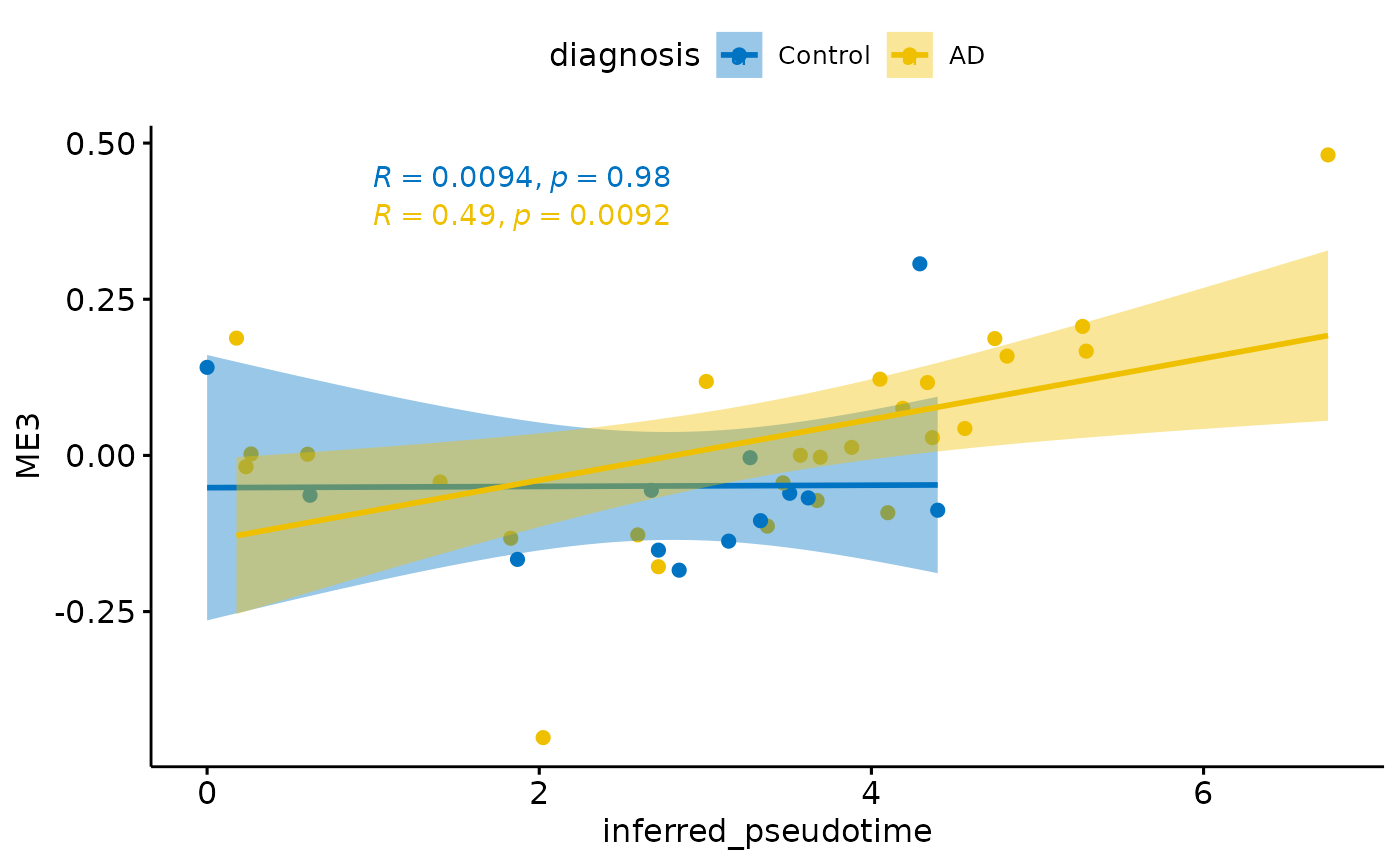
Relationship with diagnosis
for(i in colnames(modules$modules_eigen_value)){
gg=ggpubr::ggscatter(modules$metadata_modules,
x = "inferred_pseudotime",
y = paste(i),
color = "diagnosis",
palette = "jco",
add = "reg.line",
conf.int = TRUE)+
ggpubr::stat_cor(aes(color = diagnosis), label.x = 1)
plot(gg)
}
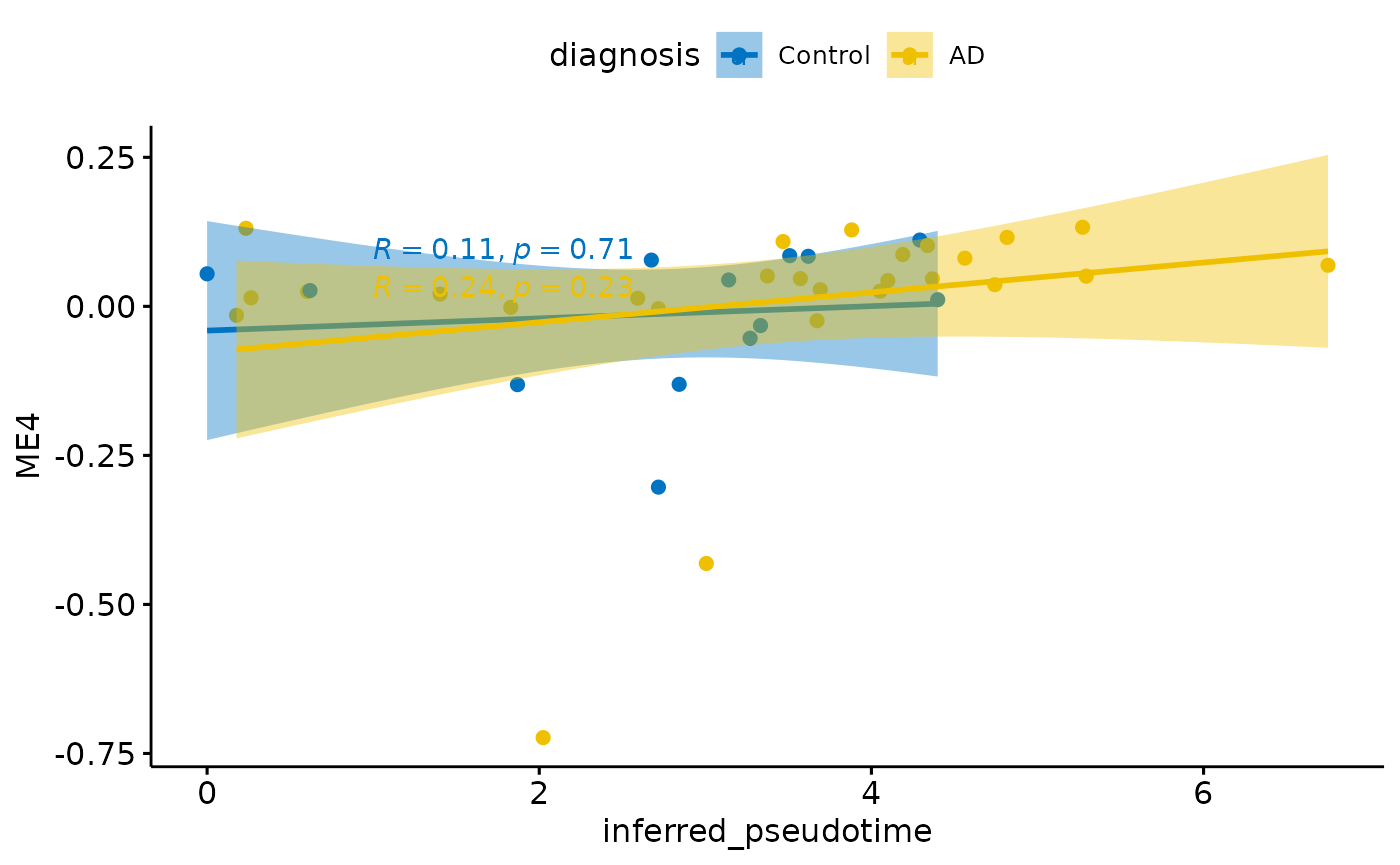
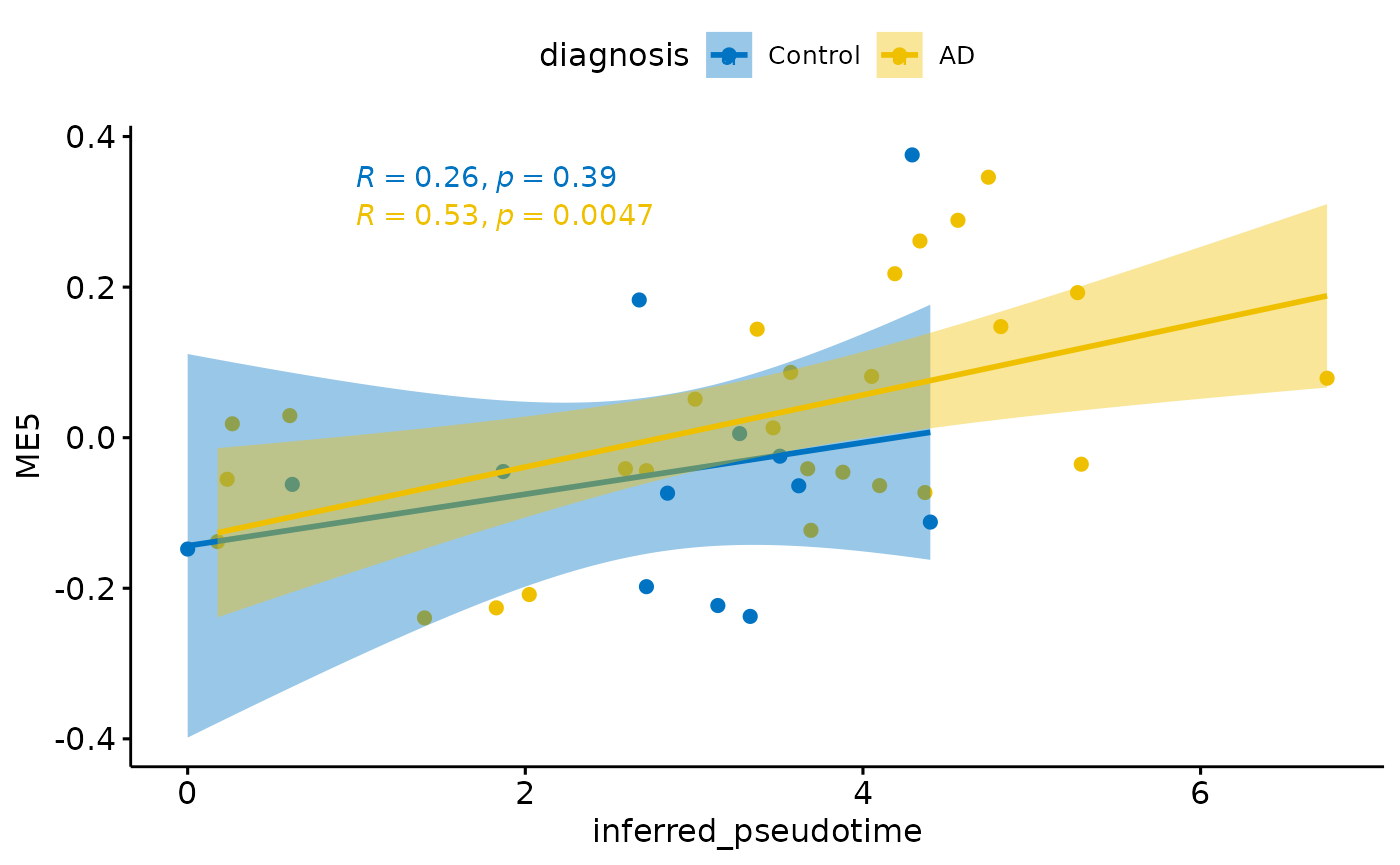
Relationship with PHF1
for(i in colnames(modules$modules_eigen_value)){
gg=ggpubr::ggscatter(modules$metadata_modules,
x = "PHF1",
y = paste(i),
palette = "jco",
add = "reg.line",
conf.int = TRUE,
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.x = 2, label.sep = "\n"))
plot(gg)
}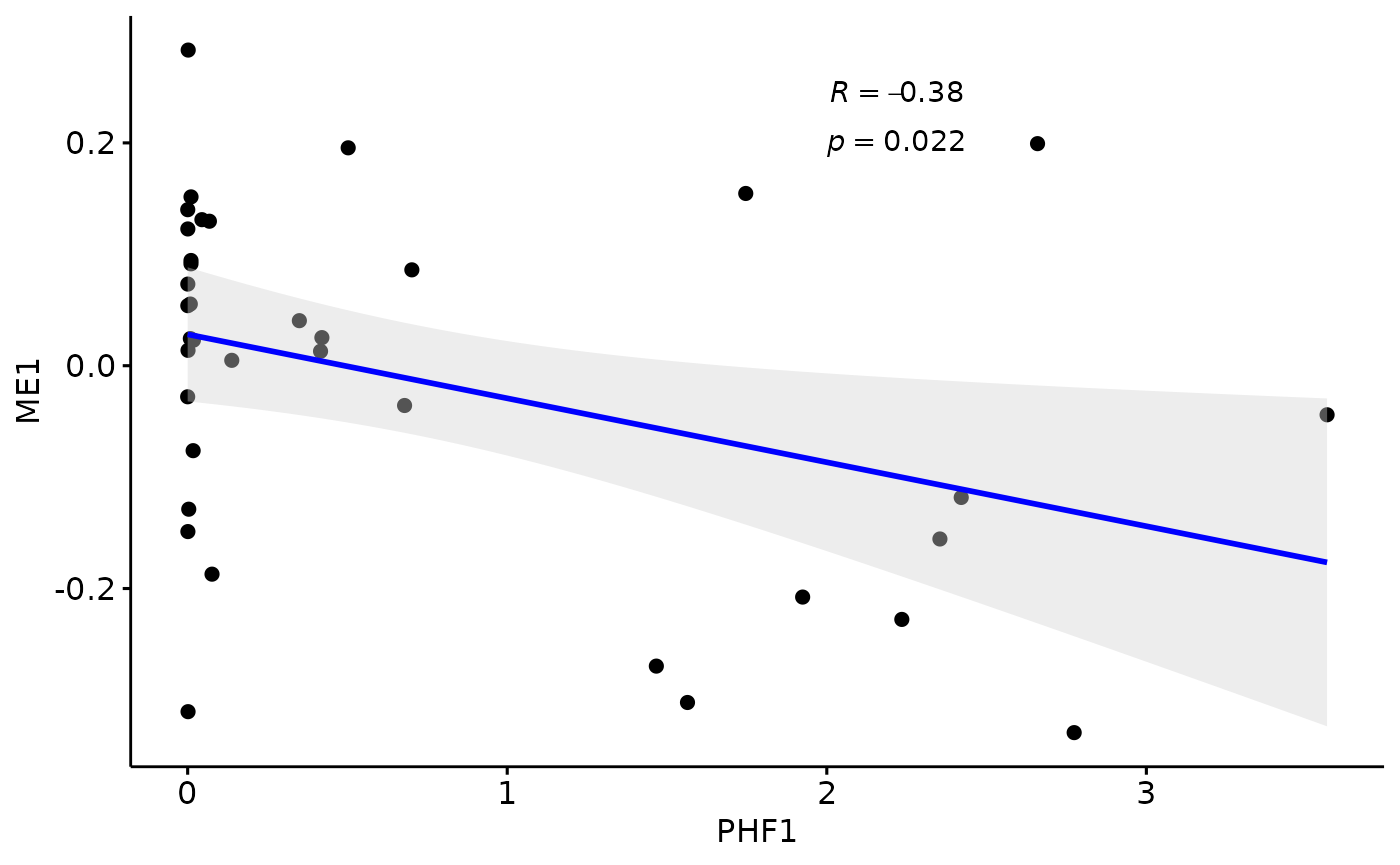
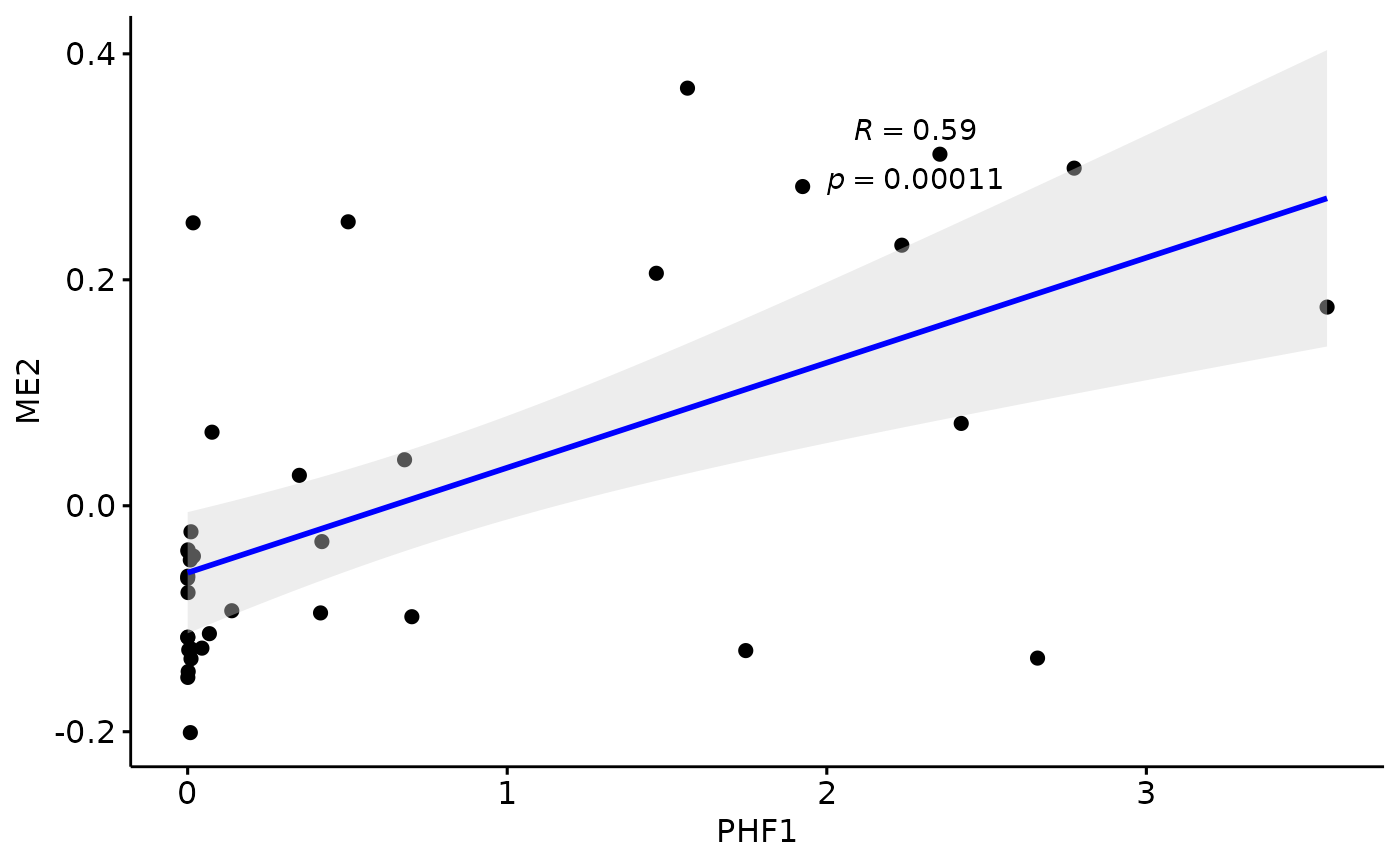
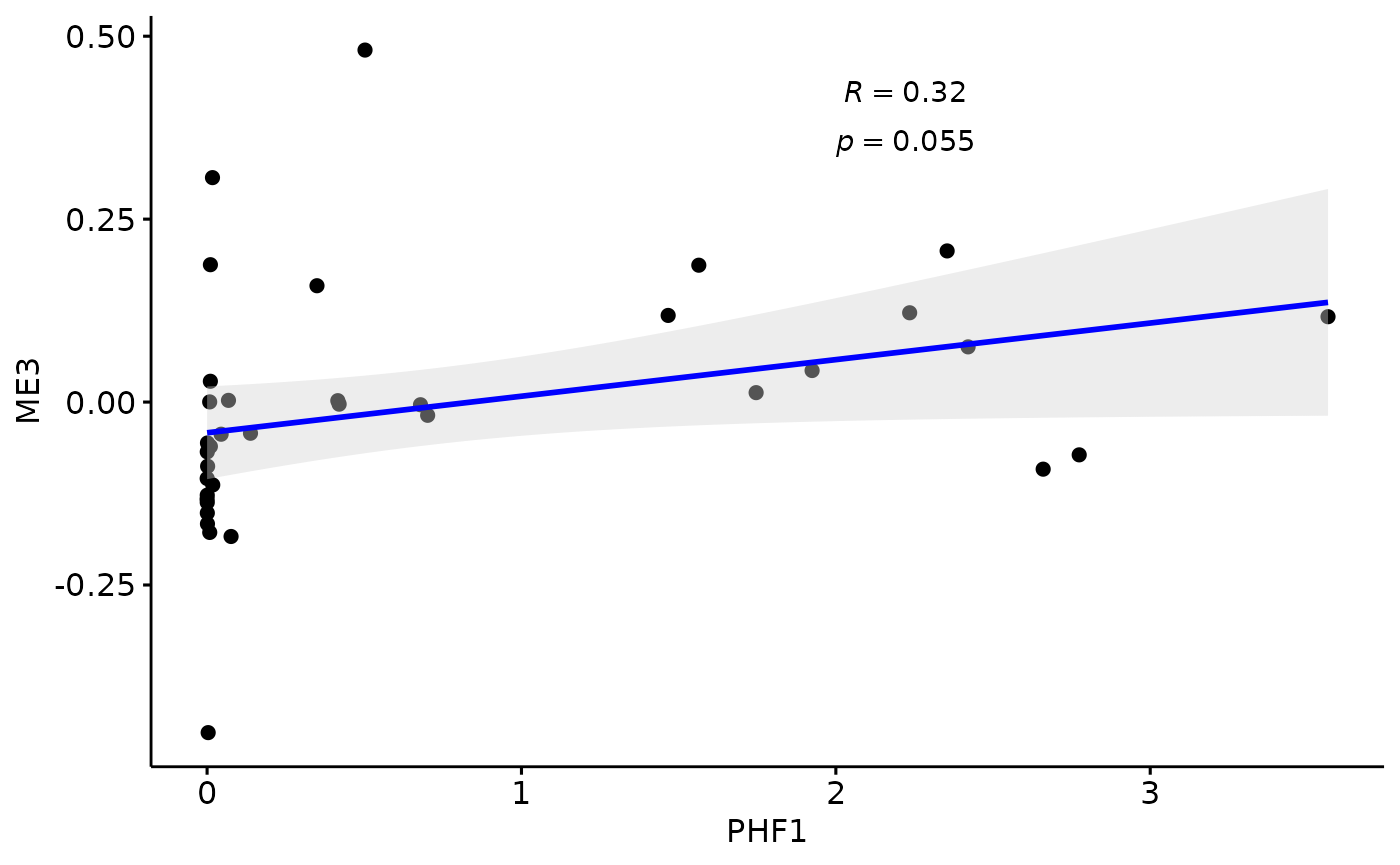
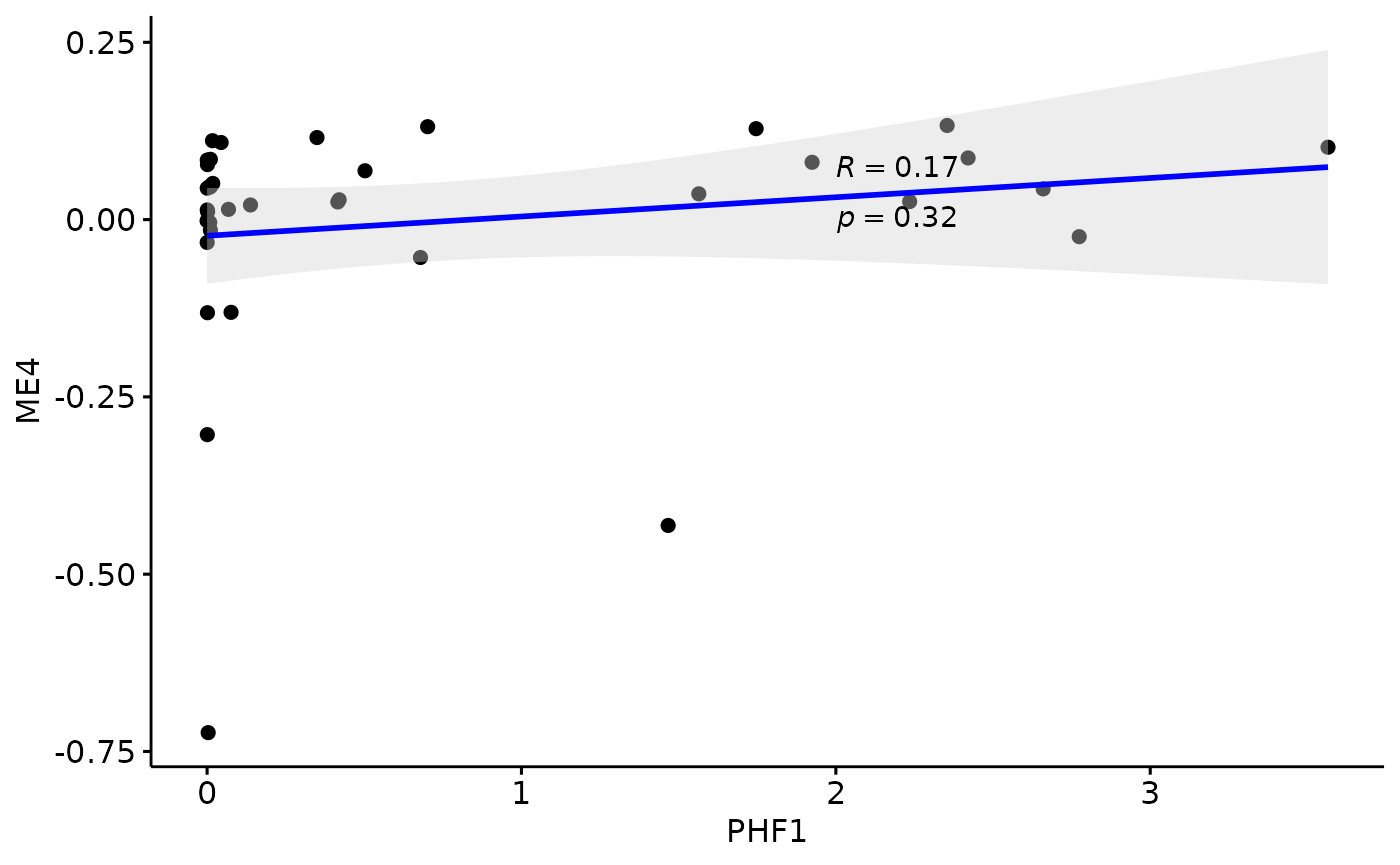
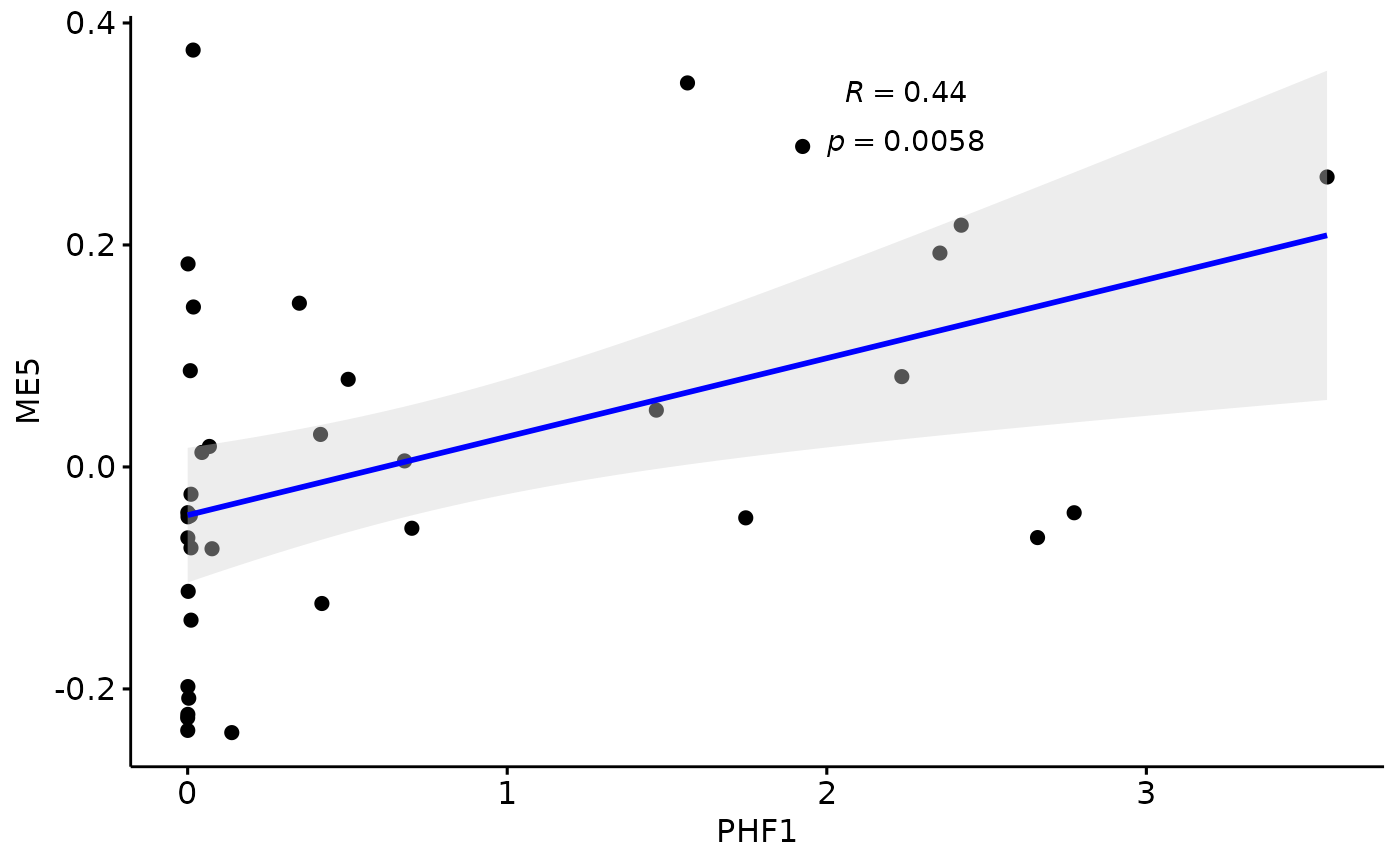
There is a stronger relationhsip between module eigen values and infered multi-omics pseudotime
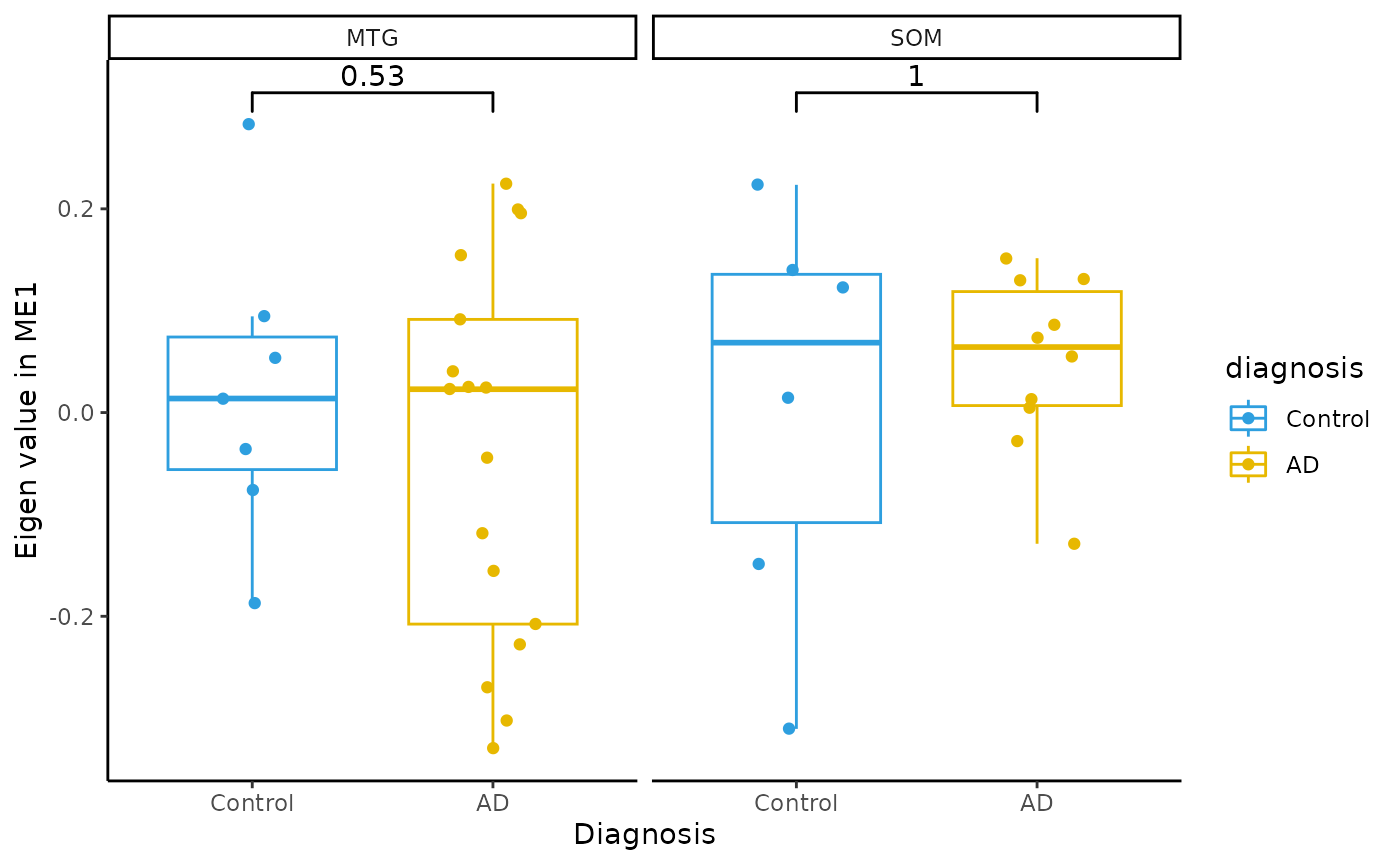
Module Eigenvalue by group boxplots
for(i in colnames(modules$modules_eigen_value)){
gg=ggpubr::ggboxplot(modules$metadata_modules,
x = "diagnosis",
y = paste(i),
add='jitter',
color="diagnosis")+
xlab('Diagnosis')+
ylab(paste('Eigen value in',i))+
theme_classic()+
ggsignif::geom_signif(comparisons = list(c('AD', 'Control')))+
scale_color_manual(values = c("#2E9FDF","#E7B800"))+
facet_wrap(~brain_region)
plot(gg)
}
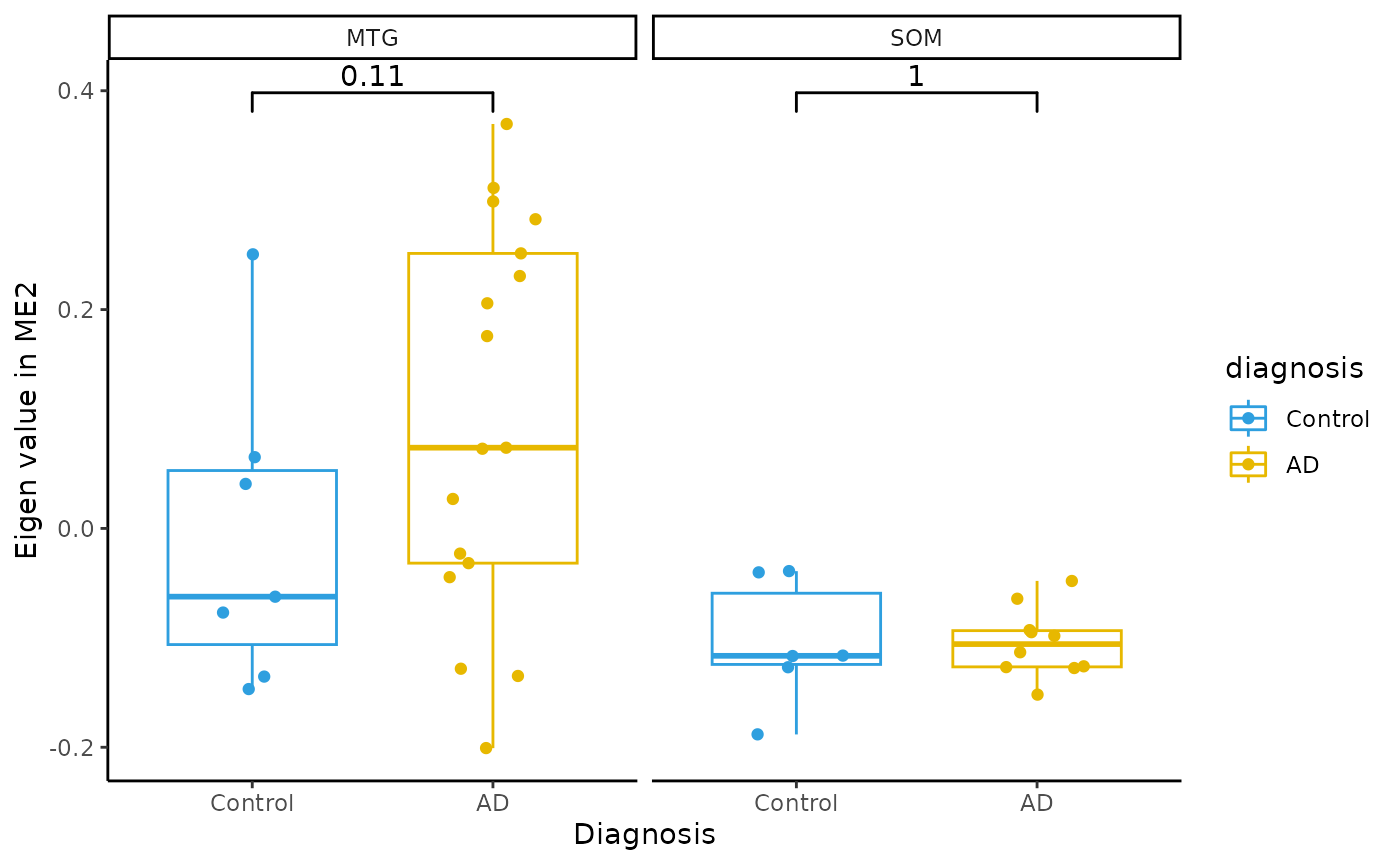
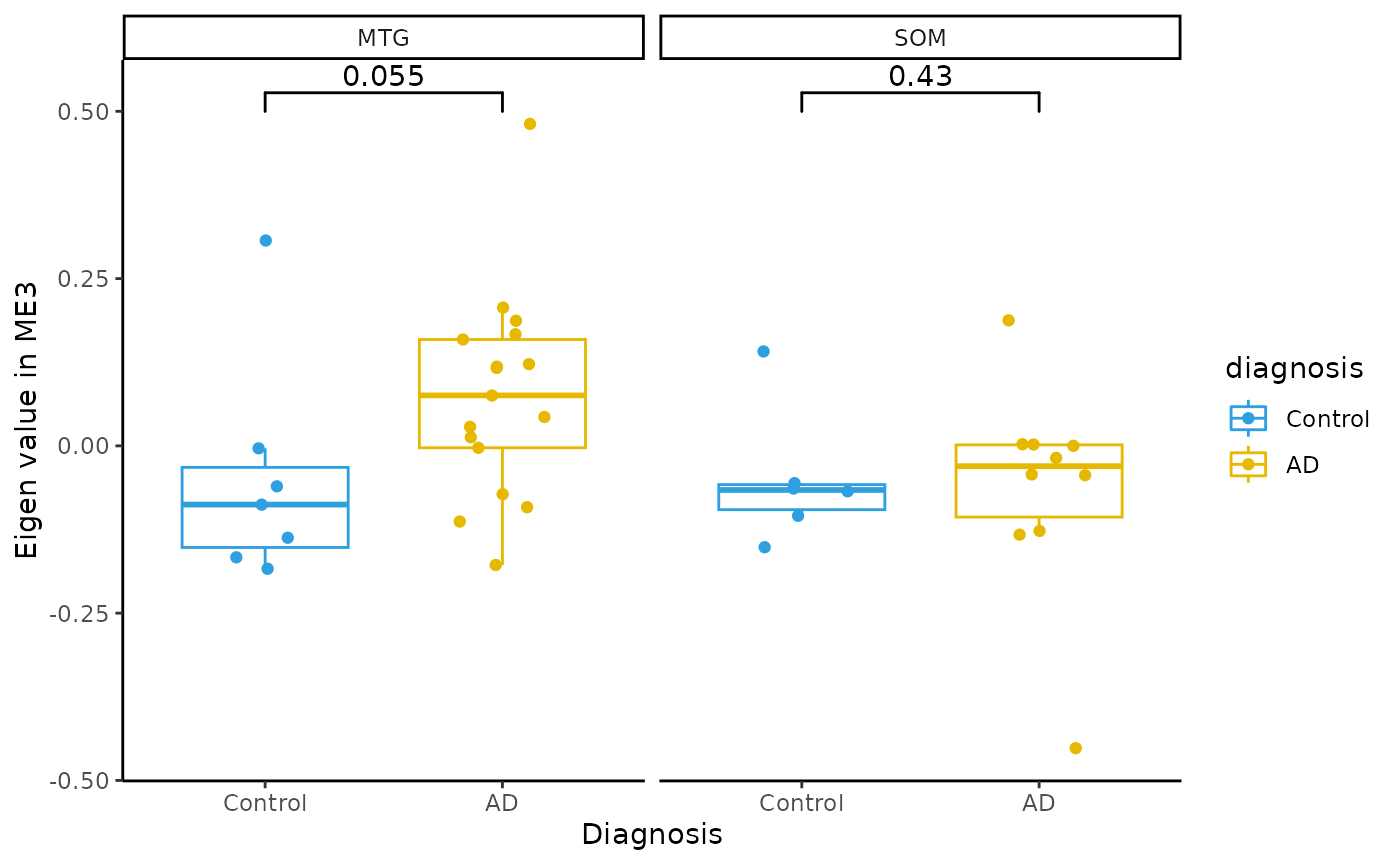
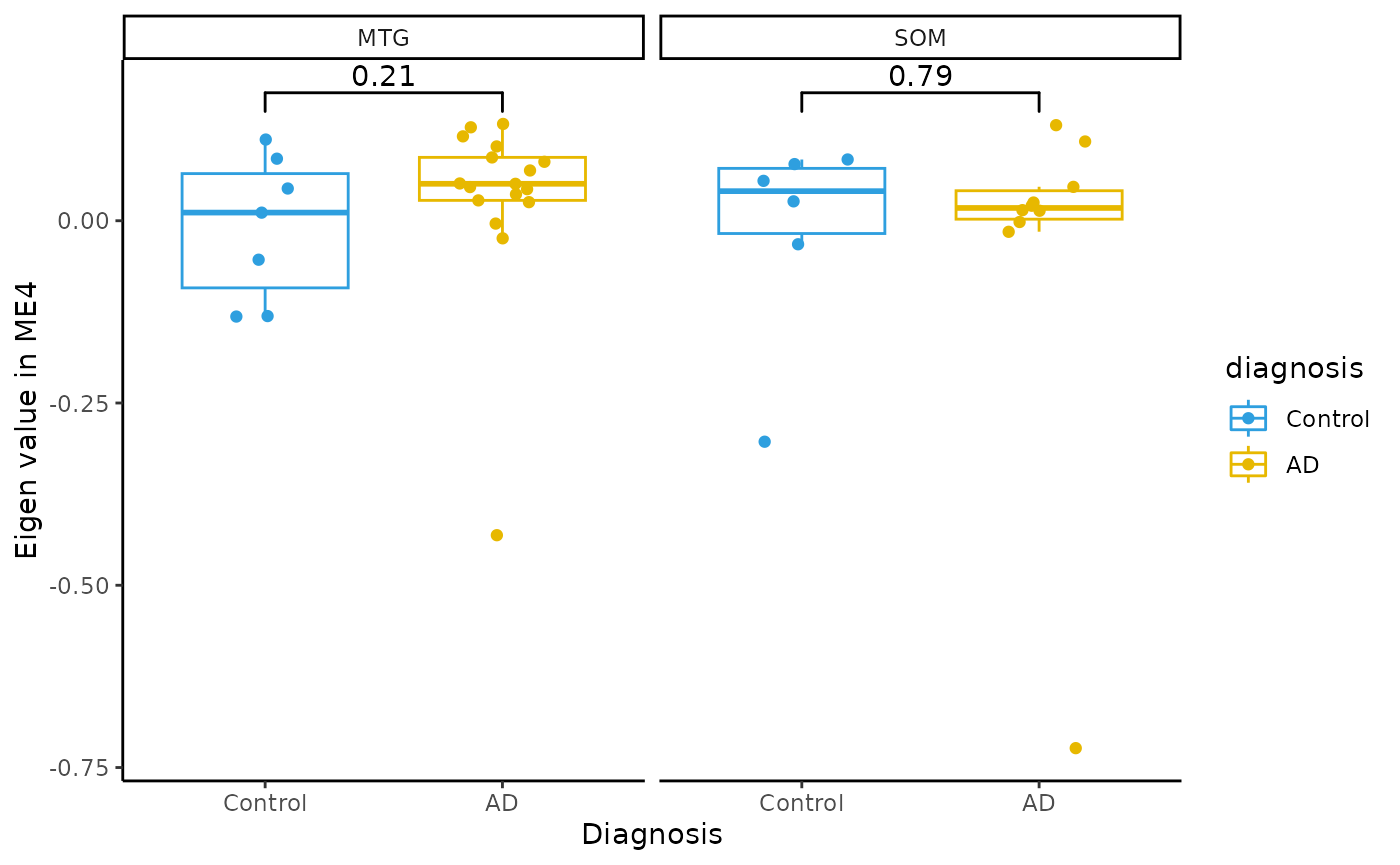
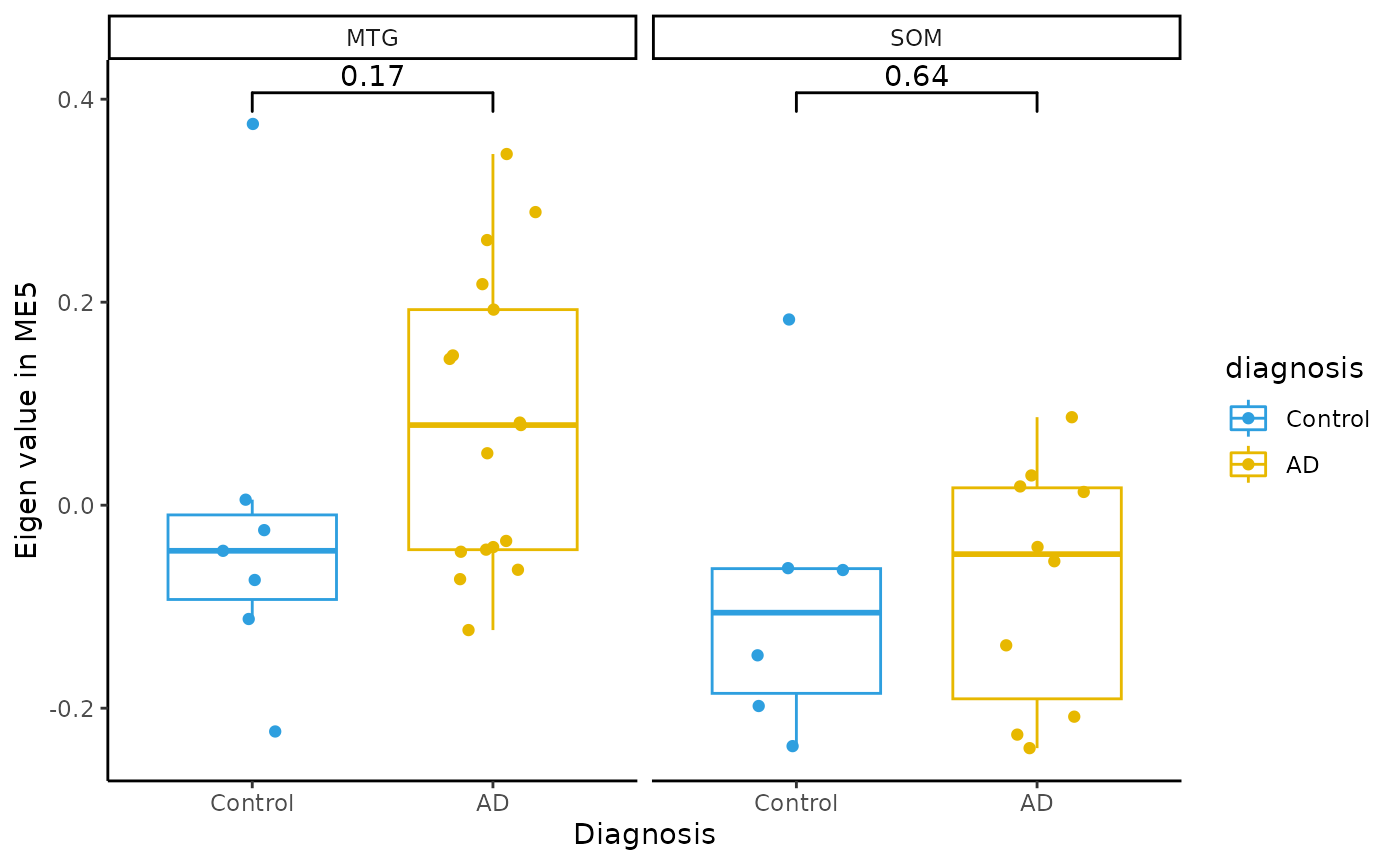
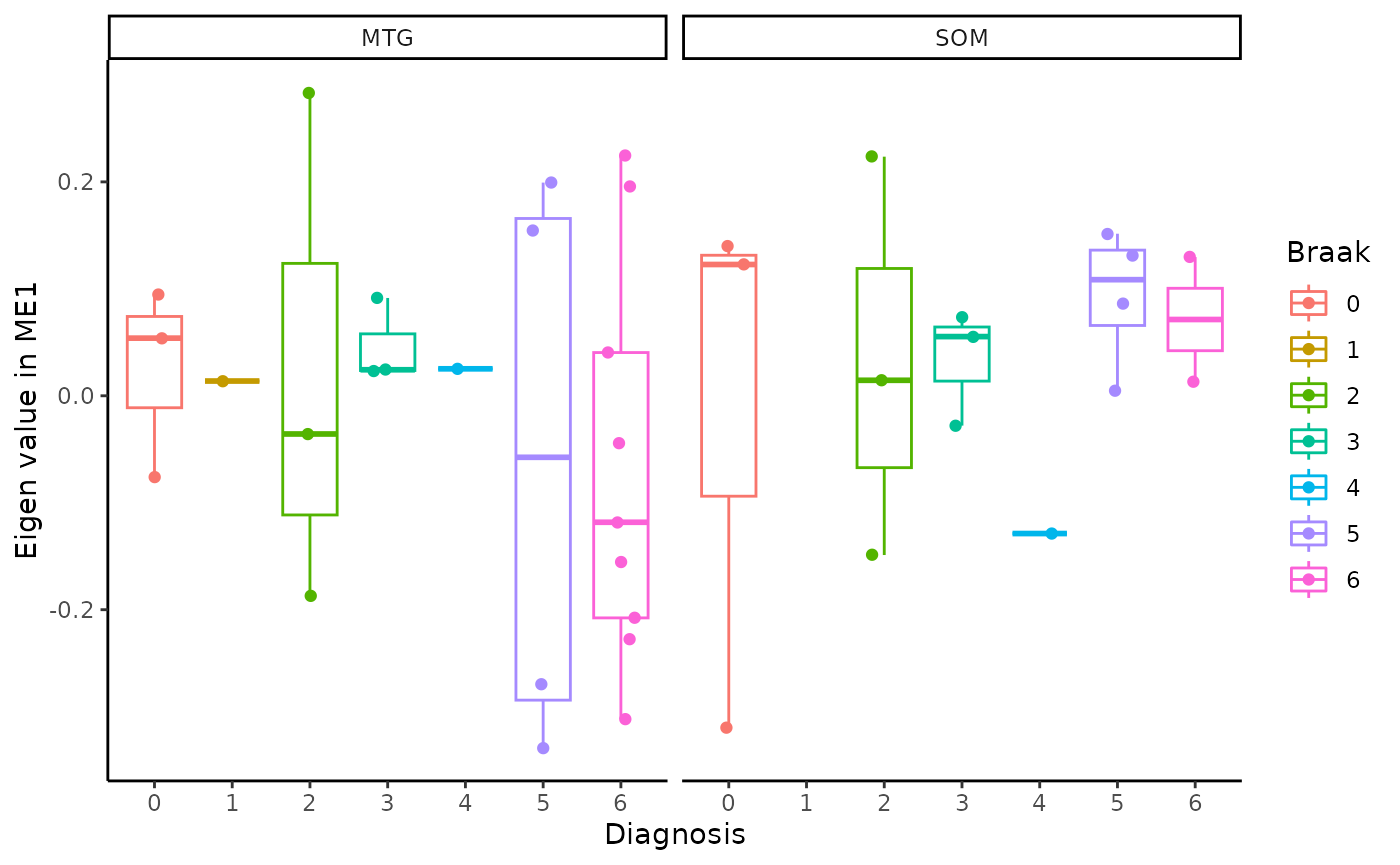
for(i in colnames(modules$modules_eigen_value)){
gg=ggpubr::ggboxplot(modules$metadata_modules,
x = "Braak",
y = paste(i),
add='jitter',
color="Braak")+
xlab('Diagnosis')+
ylab(paste('Eigen value in',i))+
theme_classic()+
facet_wrap(~brain_region)
plot(gg)
}
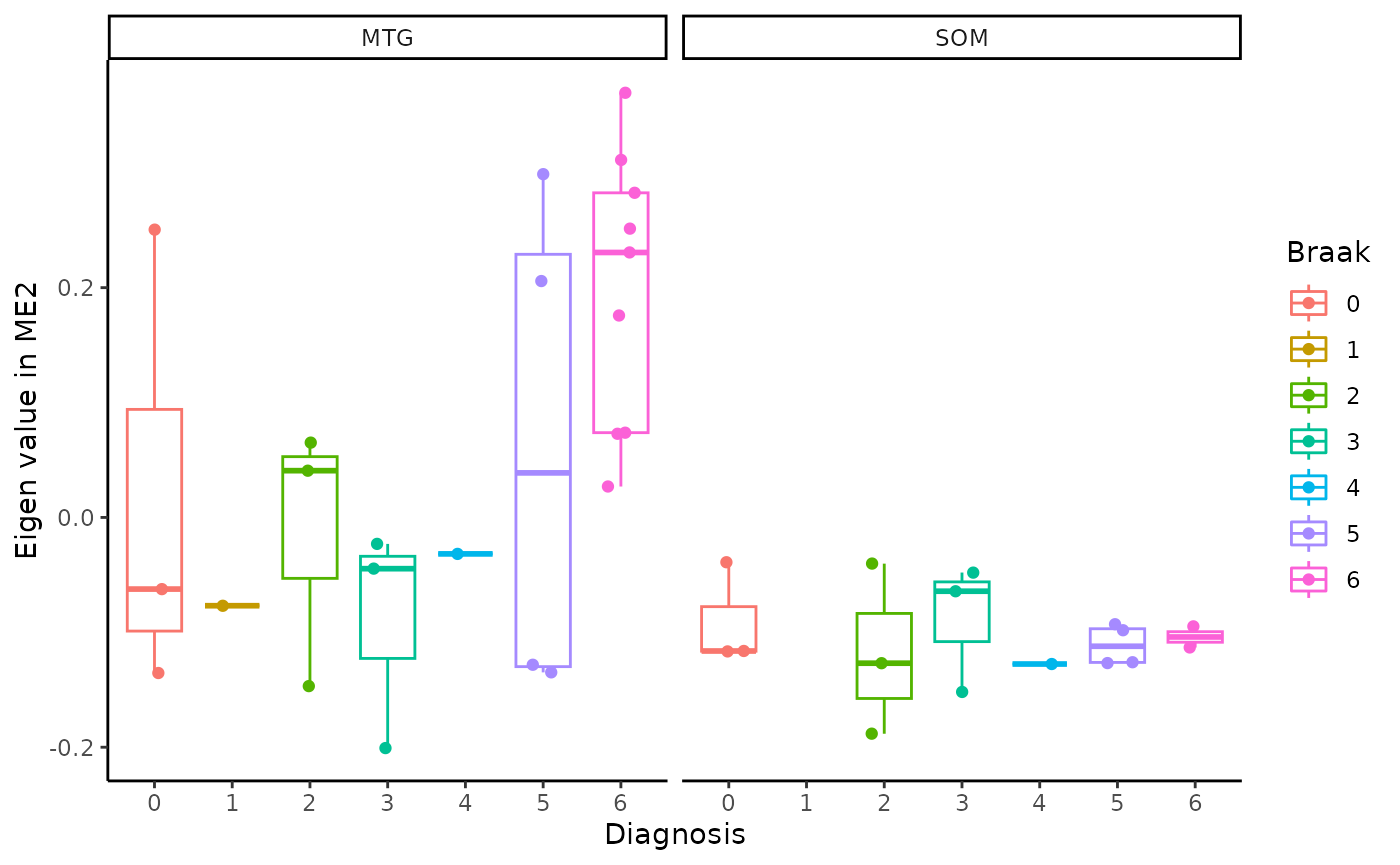
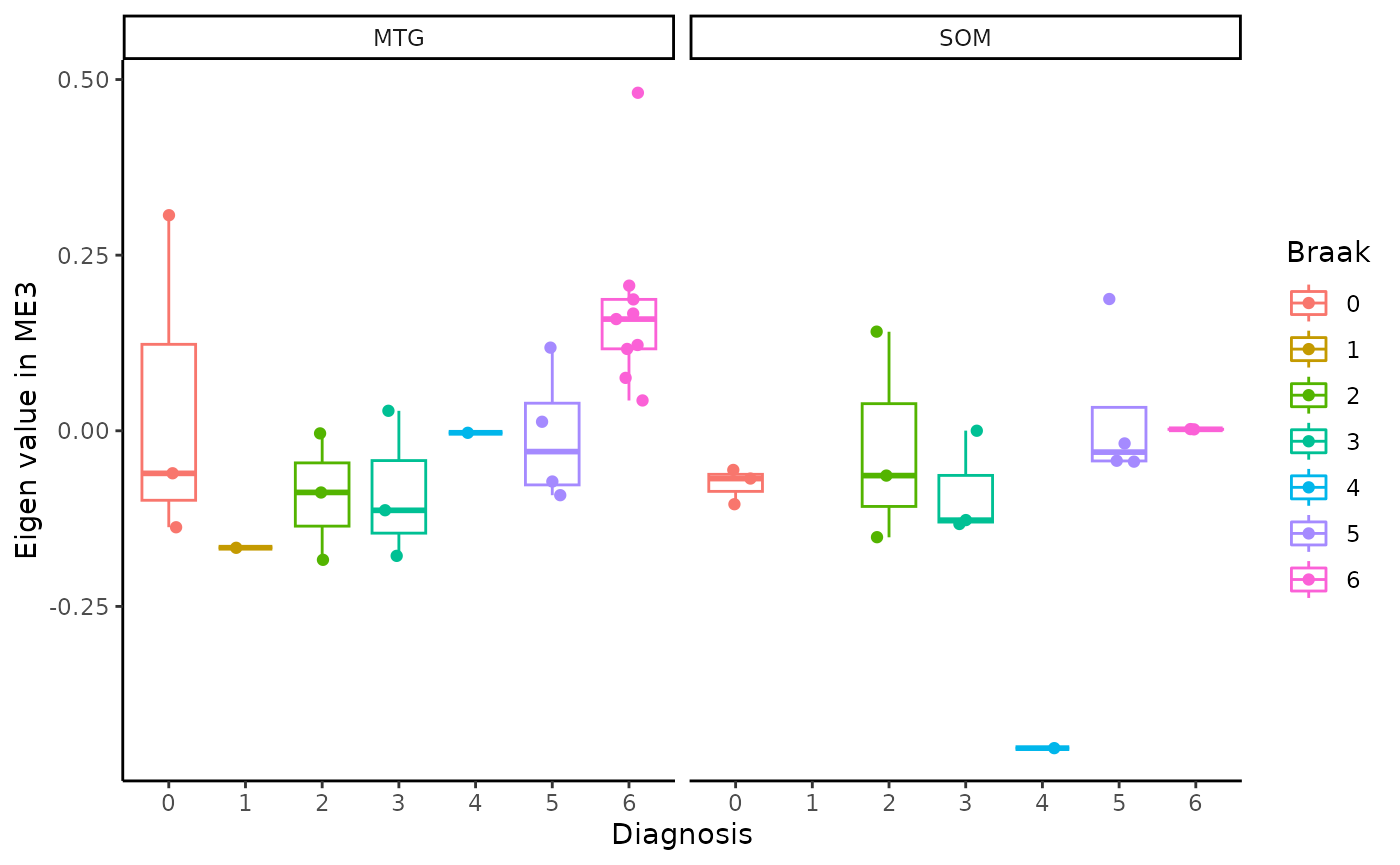
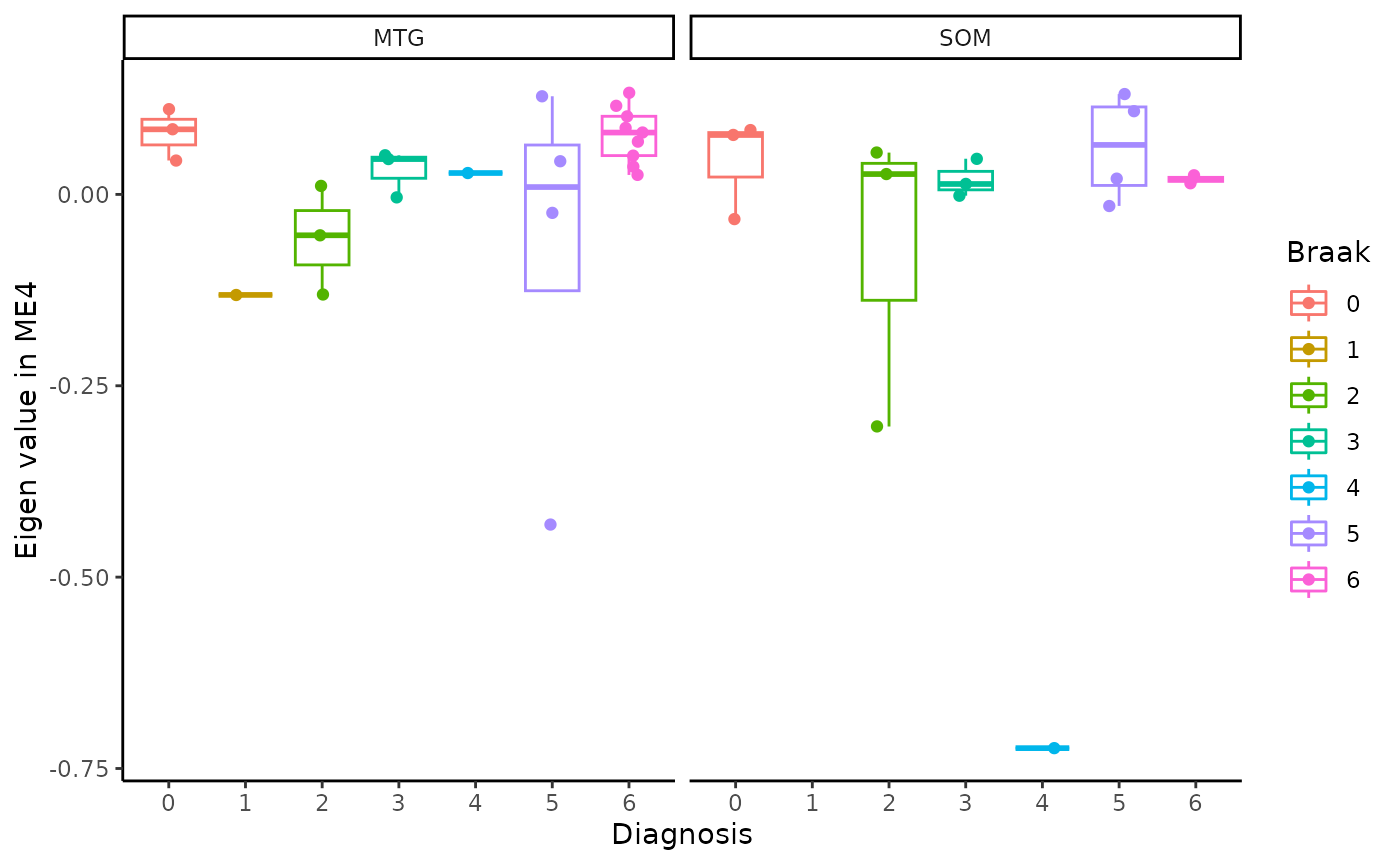
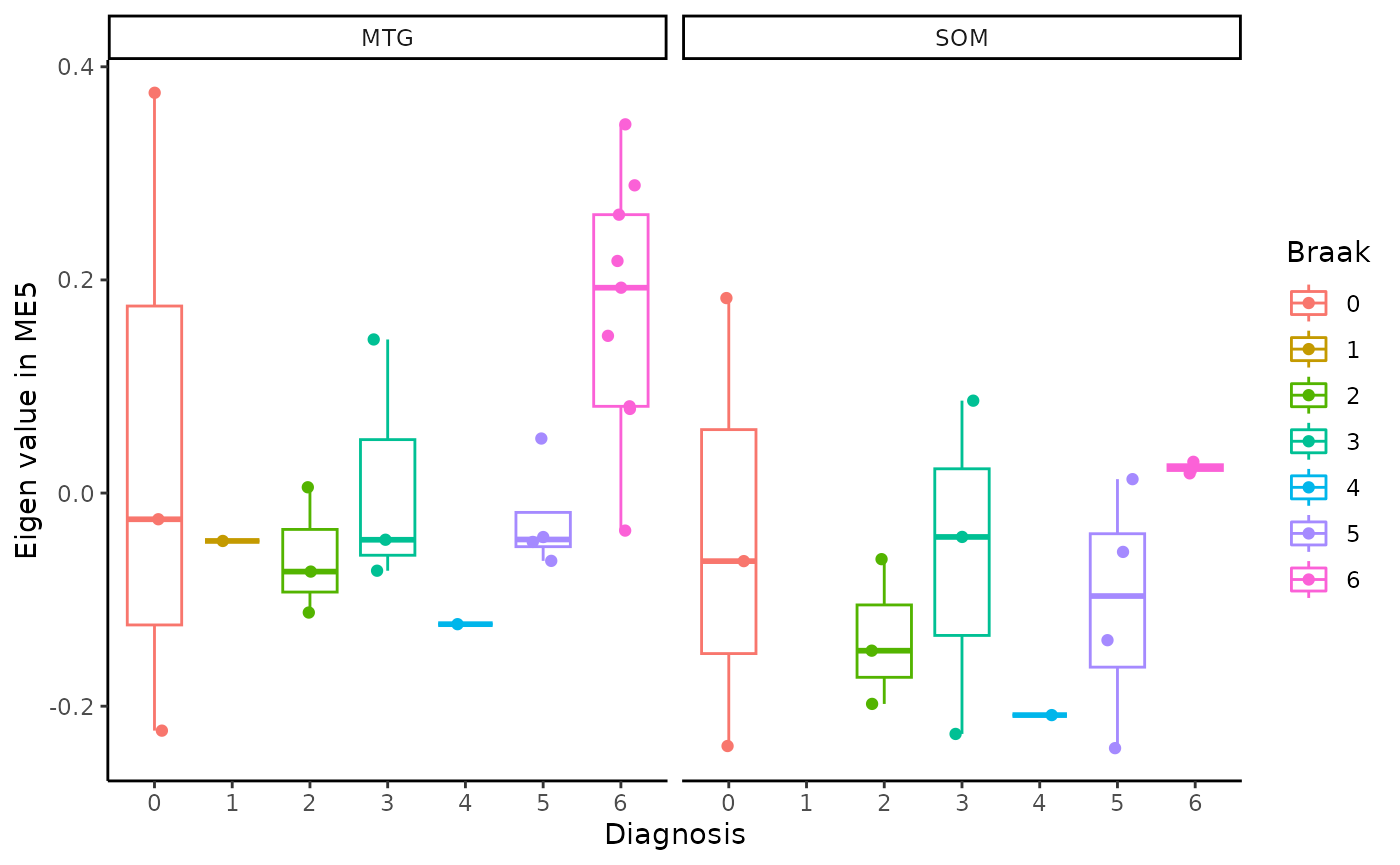
for(i in colnames(modules$modules_eigen_value)){
gg=ggpubr::ggboxplot(modules$metadata_modules,
x = "brain_region",
y = paste(i),
add='jitter',
color='brain_region')+
xlab('Diagnosis')+
ylab(paste('Eigen value in',i))+
theme_classic()+
ggsignif::geom_signif(comparisons = list(c('MTG', 'SOM')))+
scale_color_manual(values = c("#DC0000FF","darkblue"))
plot(gg)
}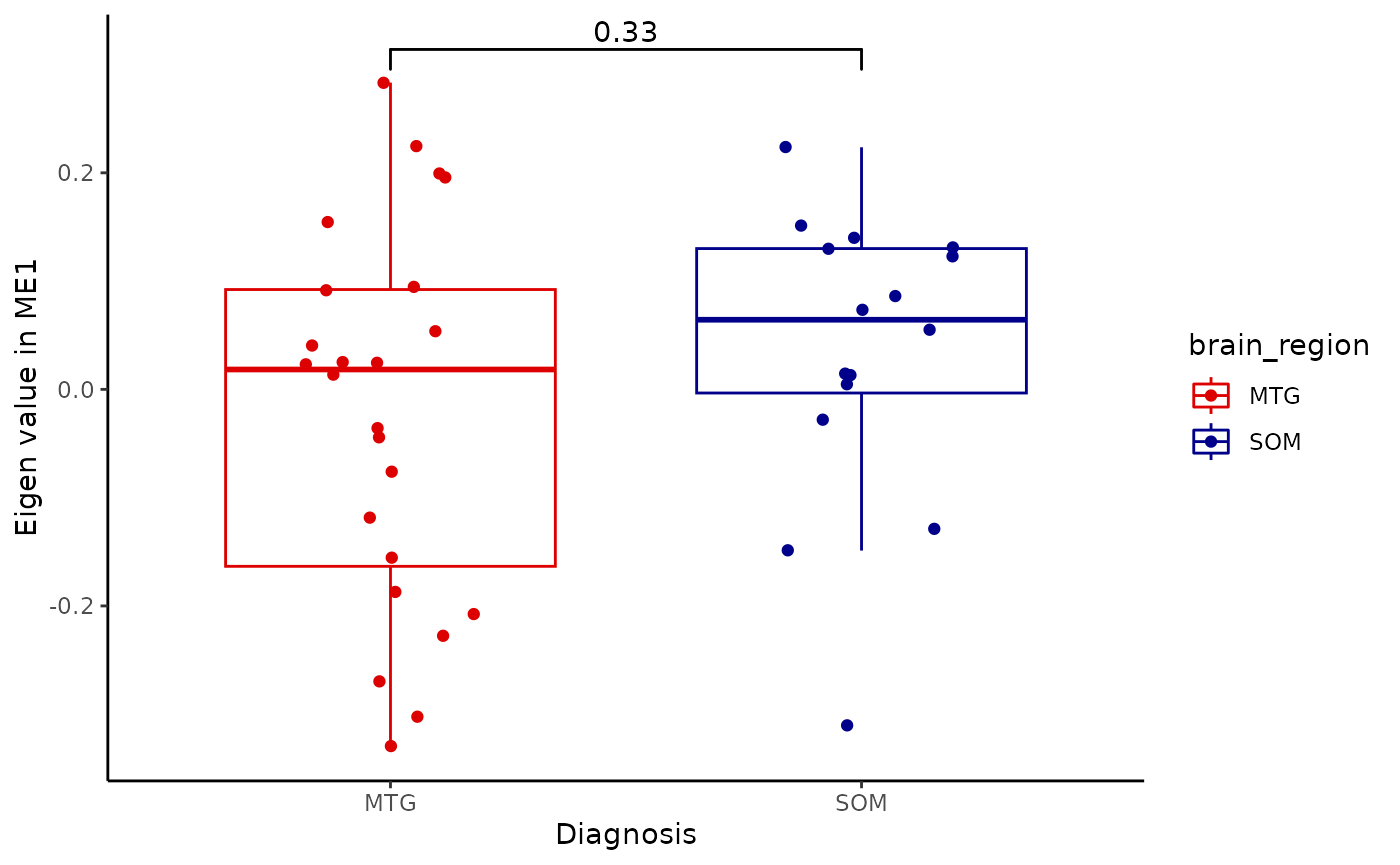
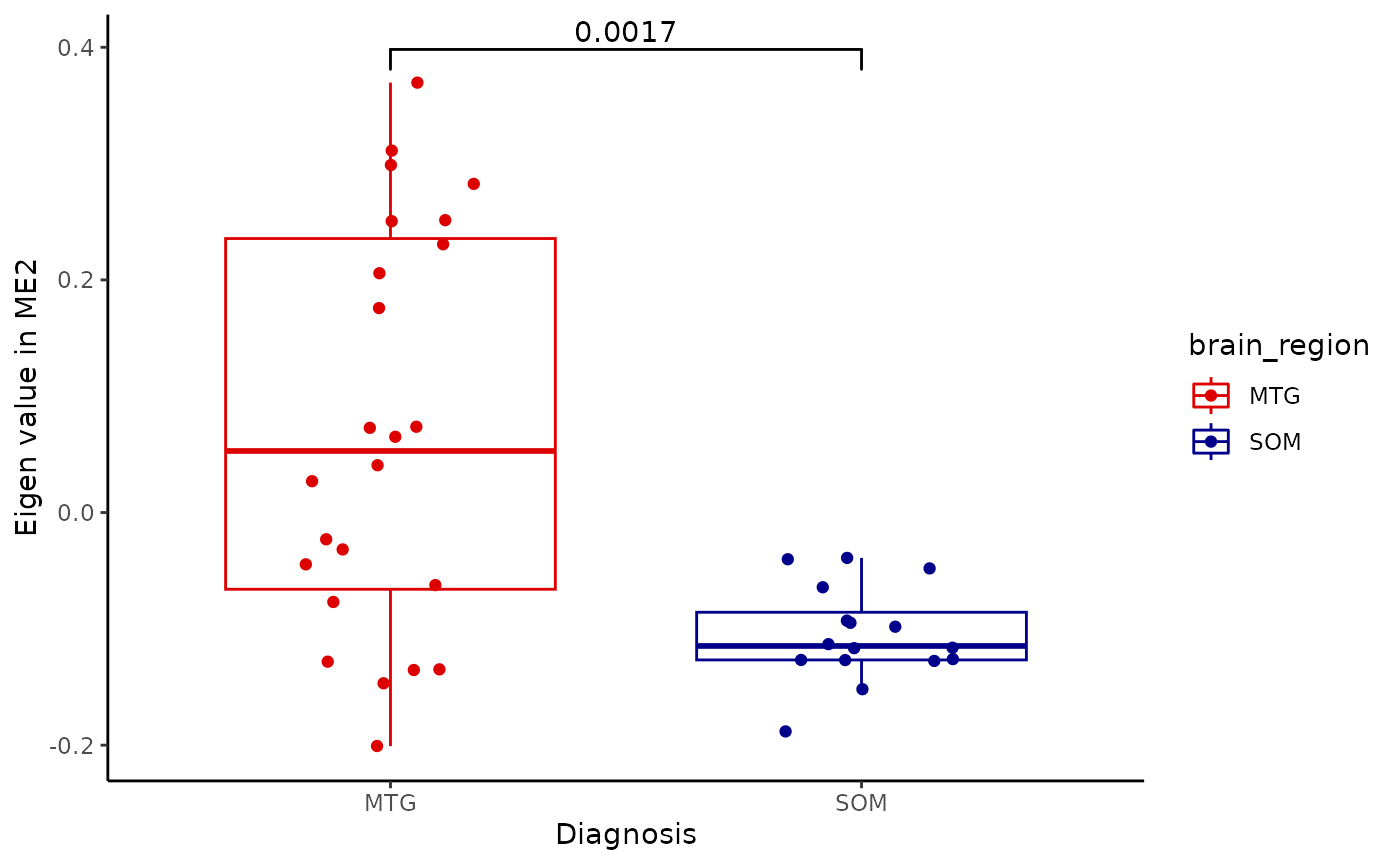
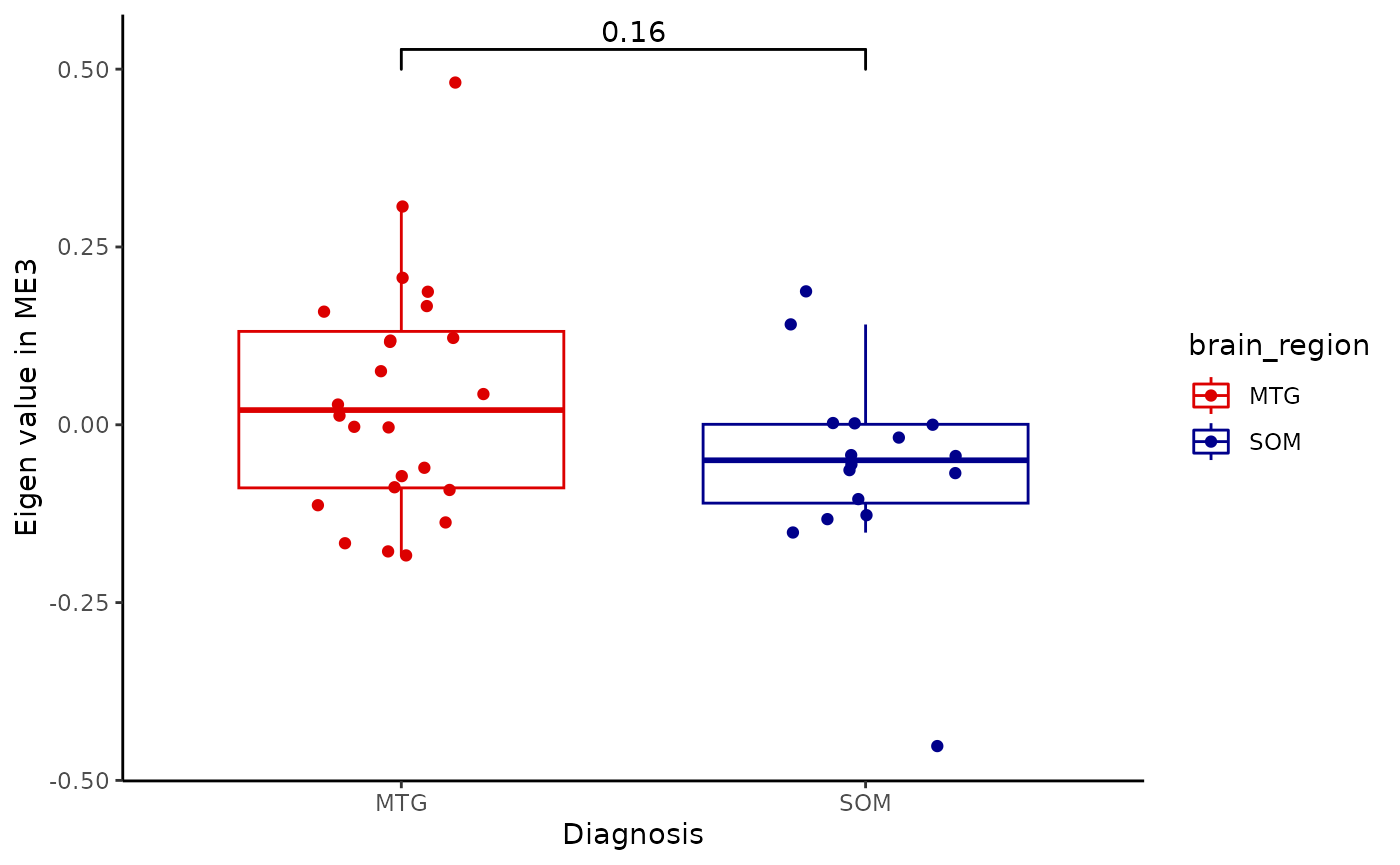
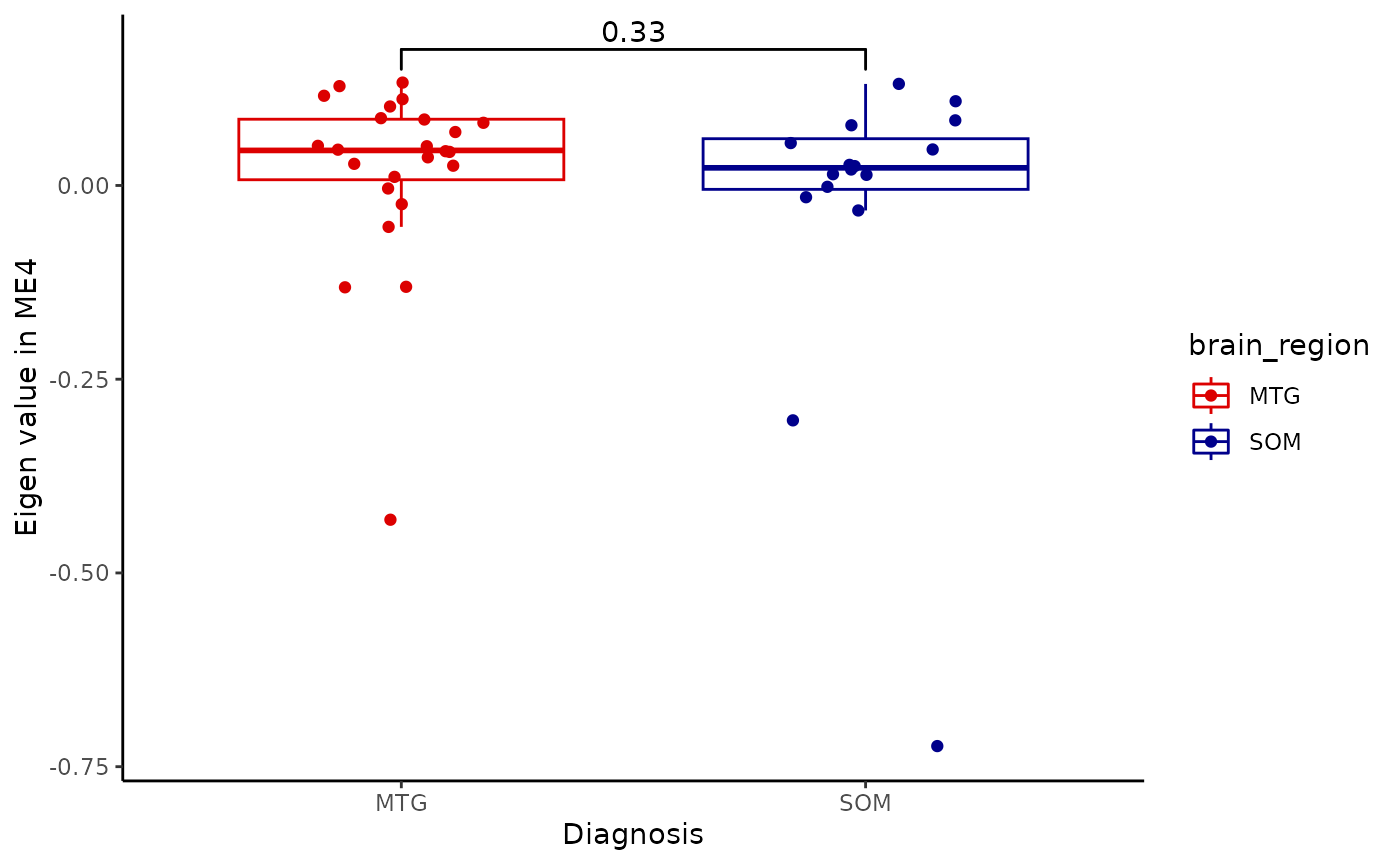
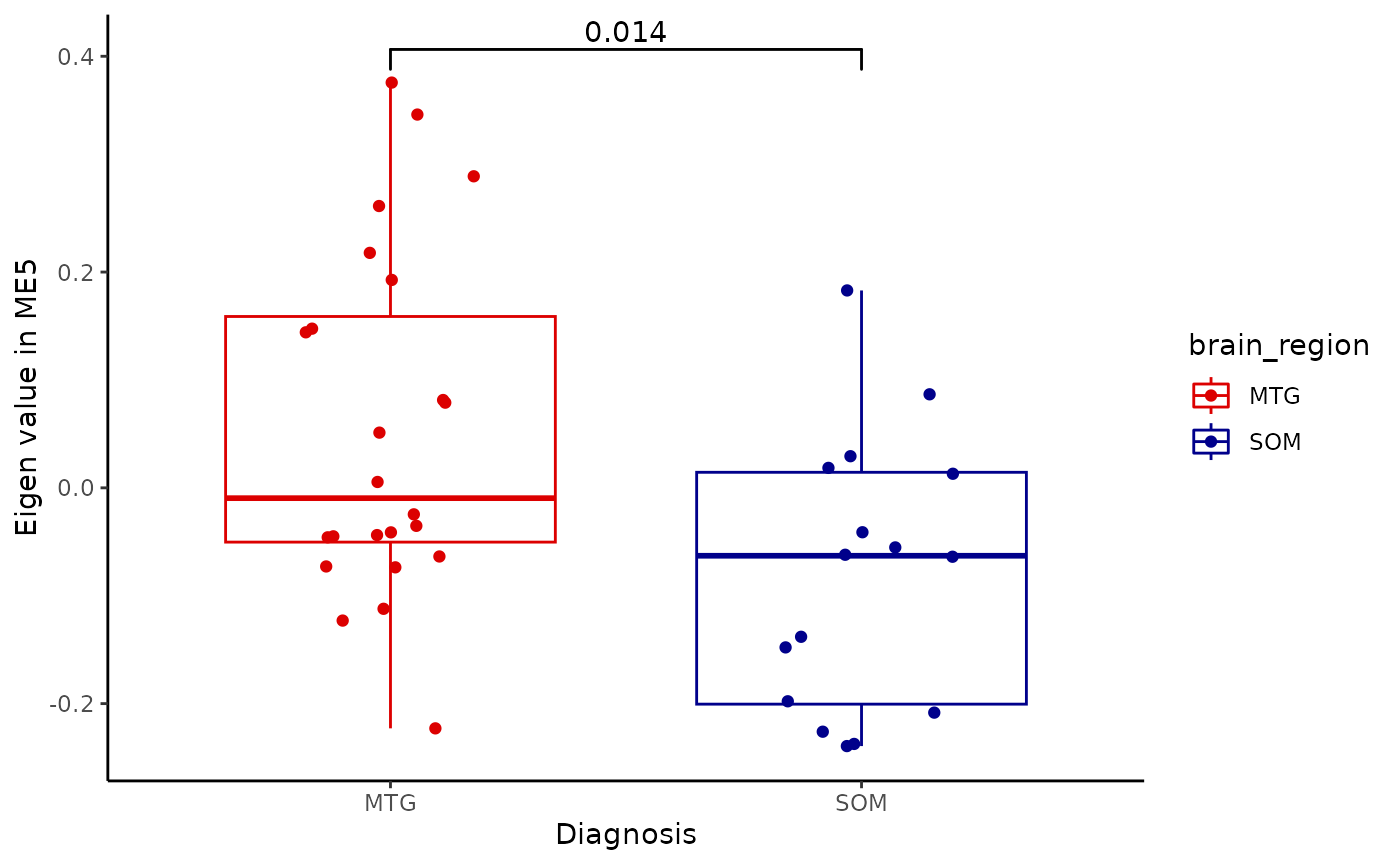
metadata=integrated_object@samples_metadata
ggplot(metadata, aes(x = brain_region, y = inferred_pseudotime,color=Braak)) +
geom_point(size = 5, col = "firebrick", alpha = 0.5,aes(color=Braak)) +
geom_line(aes(group = BBN_ID)) +
labs(x = "Brain region", y = "Inferred_pseudotime") +
theme_classic()+facet_wrap(~Braak)## `geom_line()`: Each group consists of only one observation.
## ℹ Do you need to adjust the group aesthetic?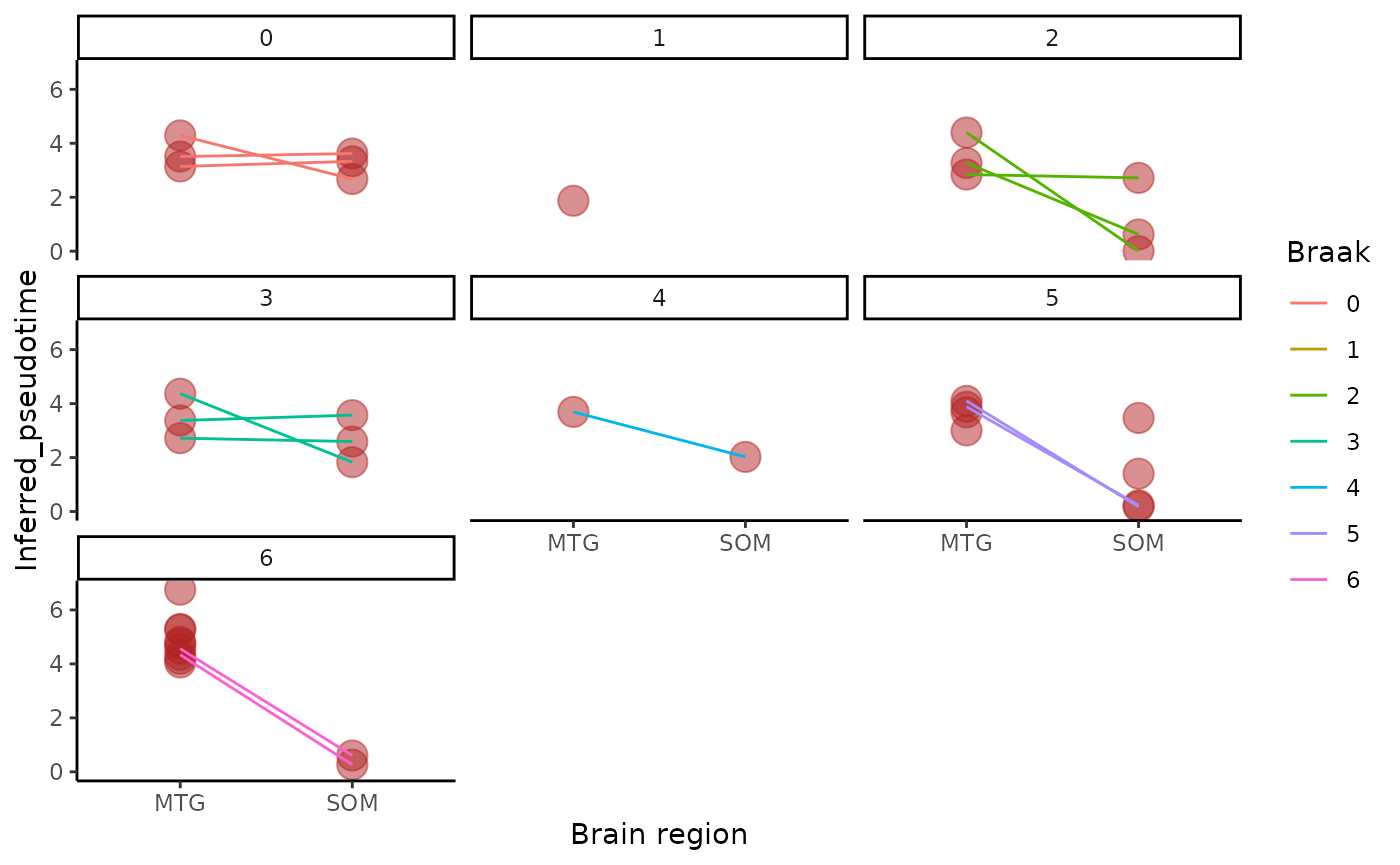
ggplot(metadata, aes(x = brain_region, y = inferred_pseudotime,color=diagnosis)) +
geom_point(size = 5, col = "firebrick", alpha = 0.5,aes(color=Braak)) +
geom_line(aes(group = BBN_ID)) +
labs(x = "Brain region", y = "Inferred_pseudotime") +
theme_classic()+facet_wrap(~diagnosis)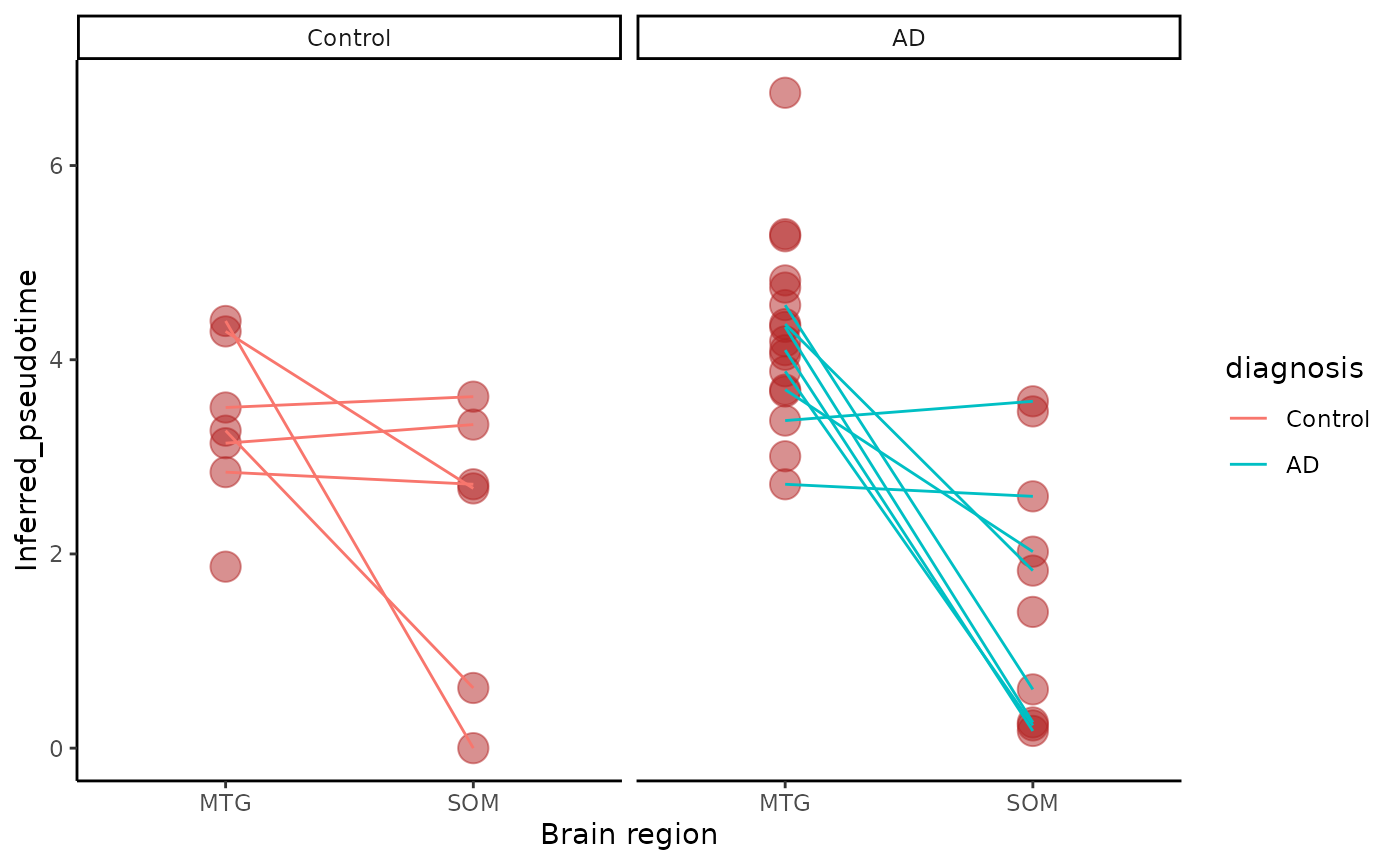
library(ggridges)
ggplot(metadata, aes(x = inferred_pseudotime, y = Braak,fill = brain_region)) +
geom_density_ridges(aes(point_shape =diagnosis, point_fill = brain_region),
jittered_points = TRUE, position = "raincloud",
alpha = 0.7, scale = 0.9, panel_scaling=FALSE
)+
xlab('Inferred pseudotime')+
ylab(paste('Braak'))+
theme_classic()+
labs(fill=NULL)## Picking joint bandwidth of 0.422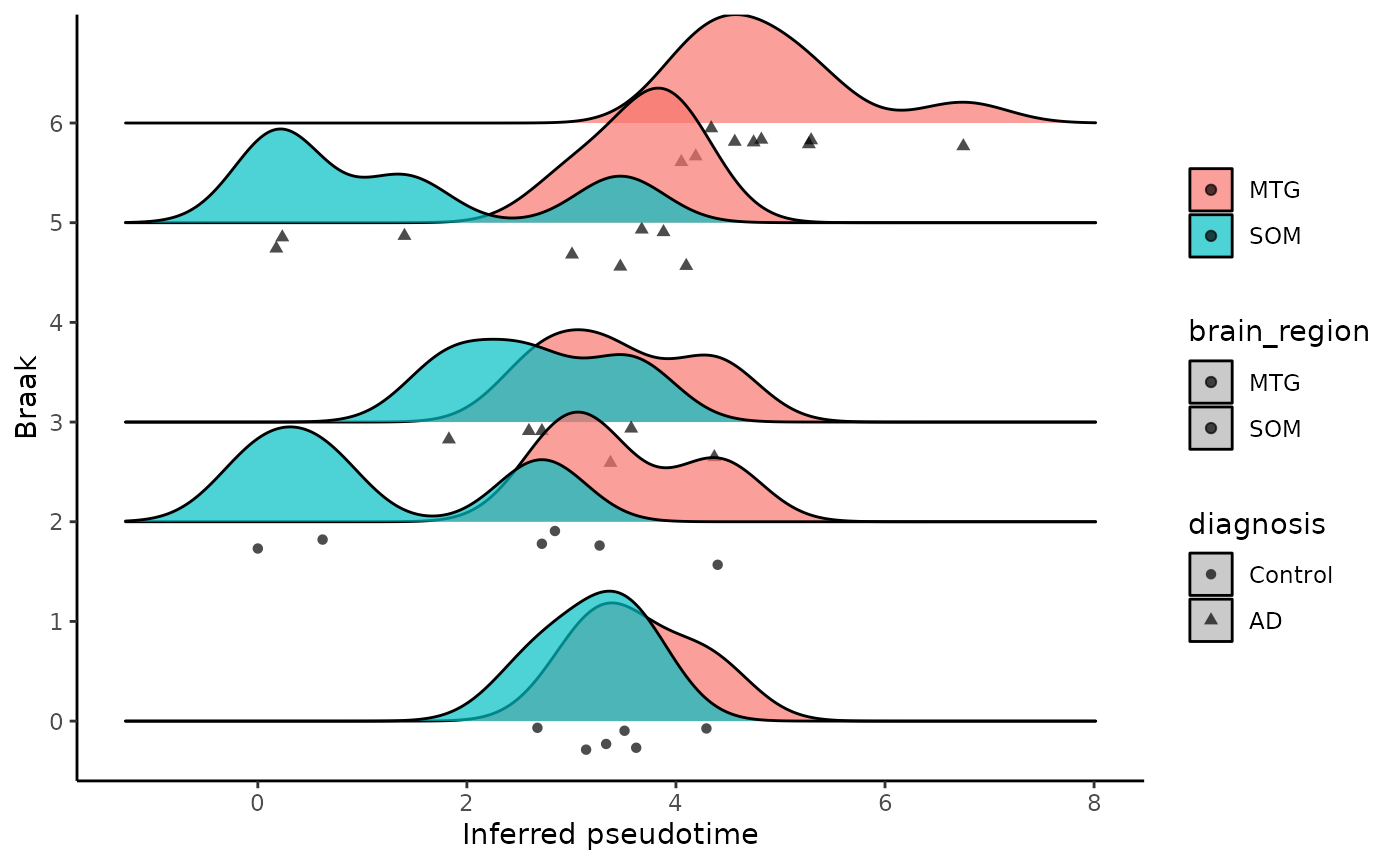
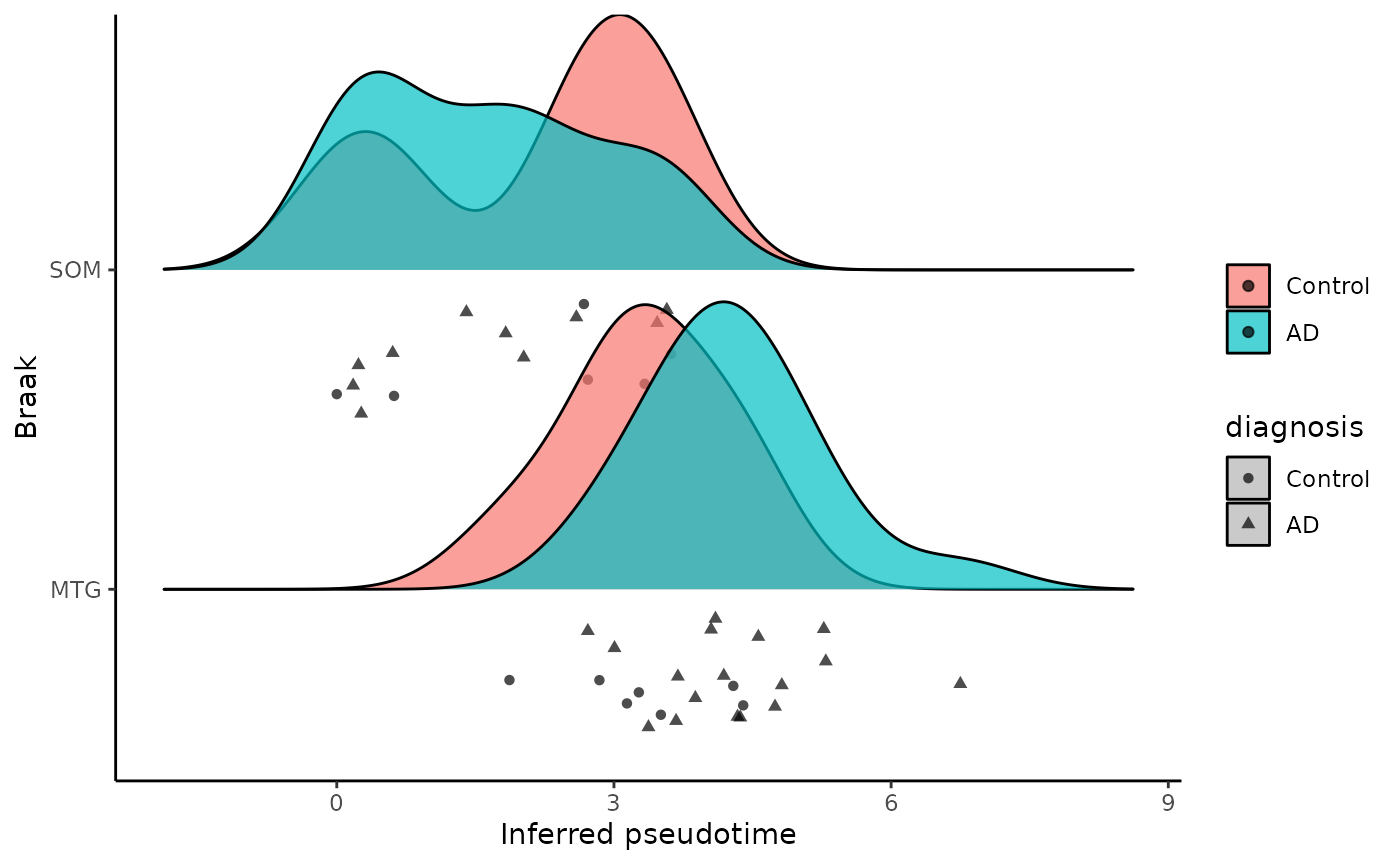
ggplot(metadata, aes(x = inferred_pseudotime, y =brain_region,fill = diagnosis)) +
geom_density_ridges(aes(point_shape =diagnosis, point_fill = diagnosis),
jittered_points = TRUE, position = "raincloud",
alpha = 0.7, scale = 0.9, panel_scaling=FALSE
)+
xlab('Inferred pseudotime')+
ylab(paste('Braak'))+
theme_classic()+
labs(fill=NULL)## Picking joint bandwidth of 0.623